



Activity modulation of various nitric oxide syntases as an approach to endothelial dysfunction therapy

- Authors: Kurkin D.V.1, Abrosimova E.E.1, Bakulin D.A.1, Kovalev N.S.1, Dubrovina M.A.1, Borisov A.V.1, Strygin A.V.1, Morkovin E.I.1, Tyurenkov I.N.1
-
Affiliations:
- Volgograd State Medical University
- Issue: Vol 10, No 2 (2022)
- Pages: 130-153
- Section: Articles
- URL: https://journals.eco-vector.com/2307-9266/article/view/111734
- DOI: https://doi.org/10.19163/2307-9266-2022-10-2-130-153
- ID: 111734
Cite item

Abstract
Nitric oxide as a therapeutic approach to the treatment of cardiovascular diseases attracted the attention of researchers at the end of the 19th century. As a vasodilator, nitric oxide may be a unique therapeutic agent for the treatment of hypertension and, as a result, renal failure and left ventricular hypertrophy.
The aim of the article is to analyze the literature data on possible ways of modulating the activity of various nitric oxide synthases as an approach to the treatment of endothelial dysfunction.
Materials and methods. When searching for materials for writing a review article, such abstract databases as PubMed, Google Scholar, e-Library, etc., were used. The search was carried out on the publications for the period from 1990 to 2021. The following words and phrases were chosen as parameters for the literature selection: nitric oxide; NO synthase; endothelial dysfunction; NO synthase activator; NO synthase inhibitor.
The following words and phrases were chosen as parameters for the literature selection:
Results. The article presents the history of the nitric oxide discovery and its biological role, the process of its biosynthesis, as well as the isoforms of its synthesizing enzymes (NOS): neuronal – nNOS, endothelial – eNOS and inducible iNOS, and their role in normal and pathological physiology. The process of NOS uncoupling (its molecular mechanisms) has been considered as the basis of endothelial dysfunction.
The examples of the pharmacological correction (BH4, arginase inhibitors, statins, resveratrol) are presented. In addition, NO synthase activators (calcium dobesilate, cavNOxin, and some NOS transcription activators), as well as non-selective (L-NMMA, 1-NNA, L-NAME, ADMA, 546C88, VAS203) and selective (L-NIO, 7-nitroindazole, aminoguanidine, L-NIL, GW273629, GW274150, cavtratin) inhibitors of nitric oxide synthasehave been described.
Conclusion. Nitric oxide synthases continue to be promising targets for the development of agents that modulate their activity to correct various pathologies. As a therapeutic approach, modulation of the nitric oxide synthase activity can be implemented to treat endothelial dysfunction, which is the cause for complications of many diseases.
Full Text
Abbreviations: 7-NI – 7-Nitroindazole; ADDP – 6-Acetyl-7,7-dimethyl-5,6,7,8-tetrahydropterin; ADMA – asymmetric dimethylarginine; AMPK – adenosine monophosphate-activated protein kinase; AP-1 – activating protein-1; AP-2 – activating protein-2; ApoE – apolipoprotein E; BH2 – dihydrobiopterin; BH3 – trihydrobiopterin; BH4 – tetrahydrobiopterin; CaD – Calcium Dobesilate; cNOS – constitutive nitric oxide synthase; CO – carbon monoxide; DDAH – dimethylarginine dimethylaminohydrolase; DHFR – dihydrofolate reductase; eNOS, NOS3 – endothelial nitric oxide synthase; ER – estrogen receptor; FGF – fibroblast growth factor; FPP – farnesyl pyrophosphate; GGPP – geranylgeranyl diphosphate; GSH – glutathione; GSSG – glutathione disulfide; GTPCH-I – guanosine triphosphate cyclohydrolase I; HUVEC – human umbilical vein endothelial cells; IFN-γ – interferon gamma; iNOS, NOS2 – inducible nitric oxide synthase; L-NAME – NG-nitro-L-arginine; L-NIO – N(5)-(1-Iminoethyl)-L-ornithine; L-NMMA – NG-monomethyl-L-arginine; L-NNA – NG-nitro-l-arginine; NF-1 – neurofibromin 1; NF-kB – nuclear factor-kB; nNOS, NOS1 – neuronal nitric oxide synthase; NO – nitric oxide (II); NOHA – NG-hydroxy-L-arginine; NOCCL – NO-cleavable cross-linker; Nrf2, NFE2L2 – nuclear factor erythroid 2, (NF-E2)-related factor 2; ODQ – 1H-[1, 2, 4] oxadiazolo[4, 3-a]quinoxalin-1-one; PCSK9 – proprotein convertase subtilisin/kexin type 9; PI3K – phosphoinositide 3-kinase; PKA – protein kinase A; PKC – protein kinase C; ROCK – Rho-associated protein kinase; ROS – reactive oxygen species; SREBP-2 – Sterol regulatory element-binding protein 2; TGF – transforming growth factor; TNFα – Tumor necrosis factor alpha; VEGF-A – vascular endothelial growth factor A; ABP – arterial blood pressure; ROI – reactive oxygen intermediate; GTP – Guanosine-5’-triphosphate; DNA – deoxyribonucleic acid; GIT – gastrointestinal tract; LDL(s) – low-density lipoproteins; LPS – lipopolysaccharide; mRNA – messenger ribonucleic acid; RA – rheumatoid arthritis; sGC – soluble guanylate cyclase; FAD – flavine adenine dinucleotide; FMN – flavin mononucleotide; cGMP – cyclic guanosine monophosphate; CNS – central nervous system.
INTRODUCTION
Nitric oxide (II) (NO) is characterized by a wide range of properties: from a toxic air pollutant to a pro-inflammatory mediator and regulator of the most important body functions (vasomotor, neurotransmitter).
NO, a colorless toxic gas, was first studied by а British chemist Joseph Priestley in 1772. A student of Theophile-Jules Peluza (who had developed nitrocellulose and other nitrosulfates), the Italian chemist Ascanio Sobrero, discovered nitroglycerin in 1847, for which he notified its ability to reproducibly cause severe headaches. Another student of Peluz, Alfred Nobel, combined this highly unstable compound with kieselgur and patented it in 1867 as a more stable commercial explosive dynamite.
In the workers involved in the production of nitroglycerin, the disappearance of angina pectoris symptoms was notified. In 1879, the British physician William Murrell proposed nitroglycerin for the angina pectoris treatment. In 1977, Ferid Murad discovered that the positive effect of nitroglycerin on vascular smooth muscles is due to the release of NO. The 1998 Nobel Prize in Physiology and Medicine was shared by Ferid Murad and Robert F. Furchgott, who, together with John Zawadzki in 1980, established the importance of the endothelial relaxing factor caused by the vasodilating action of acetylcholine.
In the late 1970s, Robert Furchgott began to investigate vasoactive effects of acetylcholine, leading to the discovery of NO.
In the early 1980s, several studies were carried out, as a result of which the chemical properties and the action mechanisms of the endothelium-dependent relaxation factor were established. The similarity between the properties of NO and the properties of the endothelium-dependent relaxation factor had been determined by 1986.
In 1987, two separate laboratories – R. Furchgott and J. Zawadzki’s, as well as Louis J. Ignarro and Salvador Moncada’s [1] – published the definitive evidence that NO is an endothelium-dependent relaxation factor [2].
The discovery of NO as an endogenously generated signaling agent of a particular importance for the cardiovascular system set off a new era in the study of biological signaling in the late 1980s and early 1990s. This discovery changed the paradigm and demonstrated that a small, freely diffusible molecule can be synthesized endogenously to act as a specific signaling factor through interactions with a receptor protein [3].
Main effects of NO
NO plays an important role in the regulation of various physiological processes that are critical for functioning of many body tissues. When NO signaling is impaired, these central tissue processes can become pathophysiological, causing acute or chronic diseases in the CNS [4–7], blood vessels [8–10], gastrointestinal tract [11, 12], skeletal muscles [13, 14] and the immune system. [15, 16].
NO biosynthesis and signal transduction in the NO-ergic system
Endogenous NO is mainly generated by enzymatic pathways, but there is also a non-enzymatic pathway (Fig. 1A). A non-enzymatic NO production involves the production of NO from nitrite in several ways, especially under conditions of acidosis (e. g, after ischemia), and occurs primarily in tissues predominantly via a nitrite reduction:
e- + 2H+ + NO2- → NO + H2O

Figure 1 – Schematic pathway for NO synthesis, including enzymatic (via NOS; main pathway) and non-enzymatic pathways (A), structure and mechanism of NOS (B, C) action. Note. A) The important sources of NO include a NOS activity and a nitrite reduction (NO2-) under hypoxic and acidic conditions. NO reacts with superoxide (O2-) to form peroxynitrite (ONOO-). Superoxide can be obtained from several sources, including a mitochondrial activity and a NADPH oxidase (NOX) activity. When protonated or combined with carbon dioxide (CO2), peroxynitrite produces a range of free radicals, including nitrogen dioxide (NO2) as well as hydroxyl (OH) and carbonate (CO3-) radicals. NO reacts with metals such as iron (FeII) to form metal nitrosyls (FeIINO) such as the one found in soluble guanylate cyclase. NO also reacts with molecular oxygen to form nitrogen dioxide and nitrogen trioxide. Together, these particles can participate in oxidative (such as thiol oxidation) reactions, nitrosation (thiol nitrosation, RSNO) and nitration (tyrosine nitration, NO2 Tyr) of biological targets; B) NOS monomers are able to transfer electrons from reduced NADPH to FAD and FMN and have a limited ability to reduce molecular oxygen to superoxide (O2-). Monomers and isolated reductase domains can bind calmodulin (CaM), which enhances the electron transfer within the reductase domain. NOS monomers are unable to bind the BH4 cofactor or the substrate L-arginine and cannot catalyze the NO production; B) In the presence of heme, NOS can form a functional dimer. Heme is required for а cross-domain electron transfer from flavins to heme of the opposite monomer. Adapted from [17(а) и 21(b и c)].
THE AIM under conditions of ischemia with acidosis, anitrite-mediated NO production approaches a maximum constitutive nitric oxide synthase (NOS) production, making this pathway an alternative in ischemia, i.e. under the conditions when the NO production from NOS is violated [17].
MATERIALS AND METHODS
When searching for materials for writing a review article, such abstract databases as PubMed, Google Scholar, e-Library, etc. were used. The search was carried out on the publications for the period from 1990 to 2021. The following words and phrases were chosen as parameters for the literature selection: nitric oxide; NO synthase; endothelial dysfunction; NO synthase activator; NO synthase inhibitor.
RESULTS AND DISCUSSION
NO synthases and their role
The enzymatic production of NO is catalyzed by various NOS isoforms through a series of redox reactions in the presence of oxygen to cleave L-arginine to L-citrulline and NO via the intermediate metabolite N-hydroxy-1-arginine (NOHA), using NADPH as an electron donor.
Isolated NOS was first isolated by Bredt and Snyder in 1990 [18]. The three major mammalian NOS enzymes had already been cloned and successfully expressed in E. coli or insect cells and had been characterized by 1998.
Three major NOS isoforms have been identified based on their different primary amino acid sequences (only 50–60% identity), tissue and cellular distribution, and a regulation mechanism. Two NOS isoforms are calcium/calmodulin-dependent and constitutively expressed (cNOS, constitutive nitric oxide synthase): NOS of the endothelial origin (NOS3, eNOS, endothelial nitric oxide synthase) and NOS of the neuronal origin (NOS1, nNOS, neuronal nitric oxide synthase), respectively. The third NOS isoform is induced by cytokines, and its activity is independent on calcium/calmodulin (NOS2, iNOS, inducible nitric oxide synthase). The individual properties of each NOS isoform are of great importance, since they are: the magnitude, duration, and cellular sites of the NO production that determine the overall physiological or pathophysiological effect [19].
While iNOS normally produces large amounts of NO and is involved in the immune defense, inflammatory responses, and the NO production by airway epithelial cells, the constitutively expressed nNOS and eNOS isoforms typically generate nanomolar NO concentrations that are important for physiological processes such as neuronal signaling, inhibition of the hemostatic system, vasodilation and control of blood pressure (BP). NO synthesized in such quantities is not rapidly oxidized either, and can directly interact with metal ions in proteins, for example, with the iron atom in the heme group of hemoglobin, myoglobin, soluble guanylate cyclase (sGC), cytochrome P450, and NOS itself [20].
All NOS isoforms use tetrahydrobiopterin (BH4, tetrahydrobiopterin), which is synthesized from guanosine triphosphate (GTP) by the enzyme GTP-cyclohydrolase-I (GTPCH-I). Other cofactors are flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), and heme. All NOS proteins are homodimers.
Functional NOS transfers electrons from NADPH from the carboxy-terminal reductase domain through the flavins FAD and FMN to heme in the N-terminal oxygenase domain. The oxygenase domain also binds the important cofactor BH4, molecular oxygen, and the substrate L-arginine. At the heme site, electrons are used to reduce and activate O2, as well as to oxidize L-arginine to L-citrulline, followed by the release of NO.
To synthesize NO, the NOS enzyme goes through two stages. At the first stage, NOS hydroxylates L-arginine to Nω-hydroxy-L-arginine, which remains bound to the enzyme. At the second stage, NOS oxidizes Nω-hydroxy-L-arginine to L-citrulline and NO.
All the NOS isoforms bind calmodulin. In nNOS and eNOS, calmodulin binding is induced by an increase in intracellular Ca2+ concentration. When calmodulin’s affinity for NOS increases, it facilitates the flow of electrons from NADPH in the reductase domain to heme in the oxygenase domain. In inducible NOS (iNOS), calmodulin binds at already extremely low intracellular Ca2+ concentrations due to the different amino acid structure of the calmodulin binding site (Fig. 1 B and C). All NOS proteins contain a zinc-thiolate cluster formed by a zinc ion that is tetrahedrally coordinated to two CysXXXXCys motifs (by one of each monomer) at the boundary of the NOS dimer. Zinc in NOS performs a structural rather than catalytic function [21].
Endothelial NO synthase
An endogenous NO production, especially in the cardiovascular system, mainly depends on the activity of the enzyme endothelial NO synthase. The eNOS enzyme was originally identified and isolated from bovine aortic endothelial cells [22, 23]. Further studies showed that several types of non-endothelial cells, such as cardiomyocytes [24], platelets [25], and neurons [26], also express eNOS.
In humans, the eNOS gene is located in the region 7q35–7q36 of chromosome 7 [27]. This gene is present as a single copy in the haploid set of the human genome.
Although eNOS is expressed constitutively, many factors regulate eNOS at transcriptional, post-transcriptional, and post-translational levels. In this regard, variations in the eNOS gene affect the regulation of the eNOS enzyme and, accordingly, the NO production [28].
Under physiological conditions, eNOS is responsible for the production of most endothelial NO, and for this reason, it plays a key role in the regulation of the cardiovascular system. This fact is constantly confirmed by clinical and experimental studies. In particular, eNOS knockout gene mice rapidly develop hypertension and have a high risk of stroke or other cardiovascular disease compared to wild-type mice. Similar disorders are also reproduced by a pharmacological inhibition of eNOS.
The regulation mechanisms of the eNOS activity are extremely complex and can be divided into two levels: genetic and protein. At the genetic level, the expression and stability of eNOS genes are associated with its activity. The eNOS promoter contains a large number of transcription factor binding sites, including activator protein-1 (AP-1), activator protein-2 (AP-2), the endothelin family, the nuclear factor-kB (NF-kB, nuclear factor-kB), and neurofibromin 1 (NF-1). These transcription factor complexes can regulate the eNOS expression; and hypoxia, lipopolysaccharide, and several cytokines can induce it.
In quiescent endothelial cells, most of the expressed eNOS attaches to caveolae, pocket-like membrane invaginations rich in cholesterol and sphingolipids. The localization of eNOS in endothelial cell caveolae inactivates the enzyme as a result of the strong and direct interaction of eNOS with caveolin-1. This protein-protein interaction inhibits the eNOS activity mainly by interfering with the calmodulin binding site. In contrast, binding of calcium-activated calmodulin to eNOS displaces caveolin-1 and promotes the eNOS activation. Calmodulin interacts with a cognate binding site on eNOS located between the oxygenase and reductase domains. This binding shifts the adjacent autoinhibitory loop and allows NADPH-dependent electron flow to the enzyme heme. However, in the absence of bound calmodulin, the electron transfer is blocked and the eNOS catalytic activity is suppressed. In addition to caveolin-1, eNOS can interact with heat shock protein 90 (Hsp90), the protein involved in a multicomponent chaperone system that is responsible for the folding of several proteins. Hsp90 is involved in eNOS folding and can allosterically modulate the enzyme by inducing conformational changes or by stabilizing the dimeric form, thereby activating eNOS. It should be notified that the eNOS activity depends on the presence of a substrate (L-arginine) and cofactors (NADPH and BH4) [28, 29].
In addition to these mechanisms, eNOS can be also activated by stimuli that do not cause a sustained increase in intracellular Ca2+ but still result in the sustained NO release. The main stimulus is a shear stress eNOS can be phosphorylated by several serine (Ser), threonine (Thr), and tyrosine (Tyr) residues. Phosphorylation of Ser1177 stimulates the flow of electrons in the reductase domain, increases the sensitivity of the enzyme to Ca2+, and represents an additional and independent mechanism of the eNOS activation [21].
Neuronal NO synthase
This type of NOS is expressed constitutively. The nNOS gene, located on chromosome 12, has a complex genomic organization. A cellular and tissue-specific regulation of nNOS is considered to be related to various transcription factors such as activator protein-2 (AP-2), transcription enhancer factor-1 (TEF-1)/MCAT binding factor, cAMP response element binding protein (CREB)/activating transcription factor/c-Fos, nuclear respiratory factor-1, Ets, nuclear factor-1 and NF-kappa B [30].
In addition, nNOS undergoes more than ten different alternative splicing options that potentially have unique structural features and catalytic properties. In the brain, the nNOSα form containing exon 2 is the main splicing variant, while nNOSµ is the predominant splicing variant in the skeletal muscle and the heart. nNOS-2, found in the mouse brain, acts as a dominant negative regulator of the nNOS activity due to the deletion in frame 105 of amino acid residues 504-608 involved in arginine binding. Both nNOSα and nNOSµ are attached to subcellular structures via the PDZ domain, while the other two forms, nNOSβ and nNOSγ, are cytoplasmic [31].
Post-translational modifications represent an additional level of the nNOS regulation activity and include phosphorylation, ubiquitination, and interactions with other structural and regulatory proteins, resulting in the formation of a functional complex.
The enzymatic activity of nNOS is modulated by phosphorylation at multiple sites and promotes the interaction between NO and other signaling pathways through various protein kinases. Phosphorylation of nNOS can occur at a variety of residues, including serine (Ser), threonine (The), and tyrosine (Tyr). This fact is associated with the activation/inactivation of the enzyme directly, or with the modulation of other regulatory domains present in nNOS.
In addition to phosphorylation, ubiquitination and sumoylation are two major post-translational regulatory mechanisms involved in levels and functions controlling of several neuronal proteins. Protein ubiquitination is a signal for the degradation of dysfunctional proteins.
In brain cells, nNOS is present in bound and soluble forms. The differential subcellular localization of nNOS explains the diversity of its functions [21, 32].
The regulation mechanisms of nNOS are similar to those of eNOS: the calcium-binding protein, calmodulin, acts as an allosteric activator and the nNOS activity is also regulated by heat shock proteins. Heat shock proteins (HSPs) comprise a family of molecular chaperones responsible for the proper folding and maturation of proteins. The Hsp90/Hsp70-based chaperone apparatus regulates the nNOS activity and turnover by modulating ligand-binding clefts [32].
The NOS of neurons contains a PDZ domain and can directly interact with similar domains of other proteins. These interactions determine the subcellular distribution and activity of the enzyme. The proteins containing the PDZ domain, such as syntrophin, PSD-95, or PSD-93, play an important role in the formation of multiprotein signaling complexes and are required for nNOS binding to various calcium sources. In skeletal muscles, nNOS is attached to the sarcolemma via syntrophin and dystrophin proteins. While in the CNS, nNOS connects to the cytoplasmic tail of NMDA-NR 2 via PSD95, which is responsible for calcium influx and the subsequent activation of nNOS. The subcellular fractionation of the brain tissue shows that about half of the nNOS in the brain is soluble, and the rest, via PSD95, is bound to the membrane fraction. PSD proteins are preferentially expressed at higher synaptic densities and interact with nNOS via PDZ domains [32, 33].
Neuronal NOS is involved in the modulation of physiological functions such as learning, memory, and neurogenesis. In the central nervous system, nNOS mediates the long-term regulation of synaptic transmission. NO formed in the CNS (under the action of nNOS) is involved in the central regulation of blood pressure [7]. In addition to the brain tissue, nNOS has been found in the spinal cord, sympathetic ganglia and adrenal glands, peripheral nerves, epithelial cells of various organs, nephrons, pancreatic islet cells, and vascular smooth muscle cells.
Inducible NO synthase
The iNOS gene is located on the 17th chromosome. The inducible isoform of NOS is mainly regulated at the level of expression. The mechanisms regulating the iNOS expression include the modulation of the promoter activity, the mRNA stability and translation, and finally protein. As with other enzymes, iNOS activity depends on the availability of the substrate. The stimuli and conditions that determine the iNOS expression depend on the functional state of the cell and its type. The inducers of the iNOS expression in one cell type may have no effect or even suppress an iNOS expression in another, even within the same organism [34].
Inducible NOS is not permanently expressed in cells, but its synthesis can be stimulated by bacterial lipopolysaccharide, pro-inflammatory cytokines (interferon-gamma, IL-1, IL-2, a tumor necrosis factor) or other agents, such as reactive oxygen species, as well as hormones that affect the synthesis of cyclic adenosine monophosphate (adrenaline, glucagon). Although iNOS is primarily identified in macrophages, the expression of the enzyme can be stimulated in practically any cell or tissue, provided the appropriate inducing agents have been identified. After the expression, iNOS is constantly active and is not regulated by intracellular Ca2+ concentrations [35].
Inducible in macrophages, NOS produces a large amount of NO, which is the basis of the cytotoxicity of these cells. Due to its affinity for protein-bound iron, NO is able to inhibit the key enzymes the catalytic centers of which contain it. These include iron-sulfur cluster-dependent enzymes (complexes I and II) involved in the mitochondrial electron transport, ribonucleotide reductase (an enzyme that limits the rate of DNA replication) and cisconitase (a key enzyme in the citric acid cycle). Higher concentrations of NO produced by induced macrophages can directly damage the DNA of the target cells and cause chains breaks and their fragmentation. The combination of these effects probably underlies the cytostatic and cytotoxic effects of NO on parasitic microorganisms and some tumor cells. Interestingly, non-immune cells can be also induced by cytokines to release enough NO to affect neighboring cells. For example, cytokine-activated endothelial cells have been shown to lyse tumor cells, while induced hepatocytes can use NO to kill malaria sporozoites. The iNOS activity is probably responsible for all these effects [21].
Unlike eNOS and nNOS, iNOS has a much higher affinity for calmodulin, so it can bind it at very low Ca2+ concentrations and thus its activity is not regulated by Ca2+/calmodulin, but mainly at the transcriptional level. The kinetic parameters of iNOS determine the catalytic activity level, which generates a higher amount of NO than eNOS and nNOS, and is less sensitive to the NO-dependent autoinhibition which contributes to the interaction of large amounts of NO with reactive oxygen species (ROS) and the generation of more toxic nitrogen species, such as nitrate [36].
The activity of expressed iNOS is regulated by various factors, including the availability of its substrate and cofactors, the interaction with other proteins, and the autoinactivation (to a lesser extent).
Several proteins interact with iNOS and regulate its activity. In the central nervous system (brain) of lipopolysaccharide (LPS) injected mice, iNOS is found complexed with a cytosolic protein called caliririn (primarily caliririn-7). Kalirin interacts with iNOS monomers, but not with iNOS dimers. In the cells expressing kalirin, most of the iNOS exists as a heterodimer with kalirin.
There is little monomeric iNOS, indicating that kalirin inhibits the iNOS activity, preventing its homodimerization. A low expression of kalirin correlates with the increased iNOS activity in the hippocampus of patients with Alzheimer’s disease. As the excess NO produced during inflammation is neurotoxic, calilirin may have a neuroprotective effect by inhibiting iNOS. In macrophages, iNOS interacts with 110 kDa protein called NOS-associated protein 110 (NAP110). NAP110 and iNOS complexes are found in the spleen and peritoneal macrophages of interferon-γ (IFN-γ) and LPS-treated mice. A co-expression of NAP110 and iNOS reduces the activity of the latter by 90%, although the amount of the iNOS protein does not change. Like kalirin, NAP110 inhibits the iNOS activity by interacting with iNOS monomers and preventing enzyme dimerization. Therefore, NAP110 may be a blocking regulator that prevents the increase in NO levels from toxicity [37].
NO can negatively regulate the iNOS activity. In stimulated mouse macrophages, the iNOS activity is increased by the addition of hemoglobin, an NO acceptor. The addition of NO donors S-nitrosoacetylpenicillamine or S-nitrosoglutathione significantly inhibits the iNOS activity, suggesting that NO may be involved in a negative feedback mechanism, but much weaker than that of other isoforms. In addition, the iNOS activity cannot be restored in activated cells without NO donors, indicating irreversible binding of NO to iNOS. A kinetic study showed that the main mechanism of iNOS autoinactivation is S-nitrosylation of the zinc tetrathiolate cluster, which causes a zinc loss, an irreversible dissociation of the iNOS dimer and a subsequent loss of its activity. Reducing agents may protect iNOS from this inactivation, suggesting that S-nitrosylation was indeed the main inactivation pathway [37].
Mitochondrial NO synthase
Although NO is a highly diffusible molecule that can cross membranes and reach mitochondria, there is also evidence that mitochondria produce NO [38] and that mitochondrial NOS exists. However, the identity of this mitochondrial NOS is still under debate [39, 40]. Several studies have shown that various tissues such as skeletal muscles [14], the heart [42], kidneys, the brain [42] and liver [41, 42], contain NOS (possibly classified as mitochondrial) with a positive immunoreactivity for antibodies against iNOS [41], eNOS and nNOS. In addition, Gao S. et. al. [43] were able to better determine the localization of eNOS on the cytoplasmic surface of the outer mitochondrial membrane in endothelial cells [43]. Elfering S.L. et. al. characterized the rat mitochondrial NOS sequence as a nNOSα isoform that can be modified by acylation, it is calcium sensitive, and has phosphorylation sites [44]. They hypothesized that these post-translational modifications might be responsible for targeting NOS at the mitochondrial inner membrane.
Confirming the results obtained by Elfering S.L. et al., Kanai A.J. et al. showed that mitochondria isolated from the hearts of mice in which the nNOSα isoform had been knocked out, did not produce NO, while eNOS-/- and iNOS-/- knocked out the mice, retaining the NO production in isolated mitochondria [45]. Lacza Z. et al. were able to detect the NO production, the NOS activity, and low levels of eNOS protein in mitochondria. However, the NOS activity was not impaired in the eNOS knockout mice [46]. In addition, the mitochondrial NO production was not affected by the nNOS and iNOS knockout mice.
Venkatakrishnan P. et al. were unable to detect the activity or expression of endothelial and neuronal NOS isoforms in rat liver mitochondria, casting doubt on the existence of mitochondrial NOS, at least in the liver [47].
Regardless of this controversy over the existence or identity of mitochondrial NOS, the recent studies show a stronger relationship between mitochondrial NOS and the respiratory chain. Parihar et al. suggested that respiratory chain complex I regulates the mitochondrial NOS activity, demonstrating the complex I-associated mitochondrial NOS activity in the isolated rat liver and brain mitochondria [48]. In addition, a study of the cardiac mitochondrial preparations demonstrated that complex I enriched fractions have higher mitochondrial NOS (nNOSα isoform), indicating that mitochondrial NOS is physically close to the complex I [49].
The conflicting results regarding the existence and identity of mtNOS can be explained by several methodological issues, which have been discussed in detail by Brookes P.S. [50]. Some of these problems include mitochondrial purity, the presence of other cell types in tissues (eg, vascular endothelial cells, inflammatory cells), the detection of the mitochondrial NO production, and the immunodetection of NOS isoforms. If mtNOS does exist despite methodological problems, the identity of mtNOS could also be related to the tissue of origin, which could explain the inconsistency in the identified isoform, for example, mtNOS could be an isoform of neurons in the brain, while iNOS could be in the brain and liver.
The existence of mtNOS can be confirmed by the relevance of NO in the control of mitochondrial respiration, since, when produced in mitochondria, NO will have a quick and easy access to its target, cytochrome oxidase, which is inhibited by NO [51]. However, it is also argued that, given the high diffusivity of NO, it is not necessary that it be produced near its site of action [52]. For example, NO can be produced by eNOS in vascular endothelial cells or by eNOS located on the surface of the cytosol of the outer mitochondrial membrane, then diffuse through other compartments reaching the respiratory chain [52].
Nitric oxide receptors
The most important physiological signaling pathway stimulated by NO, is the activation of soluble guanylate cyclase and the formation of cyclic guanosine monophosphate (cGMP). Soluble guanylate cyclase (sGC) is a key protein in the regulation of many physiological functions in mammals. This is a major NO receptor that plays a central role in the BP regulation, wound healing, memory formation, and other key physiological processes. The involvement of rHC is oftener and oftener found in various pathophysiological processes [53].
Since the rate constant of the NO association with heme centers is usually very high compared to other potential targets, even low, nanomolar, concentrations of NO are sufficient to activate sGCs. Rapid reactions between NO and targets with such kinetics limit alternative signaling processes that may otherwise be detrimental to cell functions. Upon the NO stimulation, sGC generates cyclic guanosine monophosphate from guanosine-5’triphosphate and activates protein kinase G-dependent signaling, required for various signaling cascades in several cell types. It is well known that dysregulation of sGC signaling leads to dysfunction of the cardiovascular, nervous, and gastrointestinal systems [54].
The physiologically significant mammalian sGC heterodimer consists of α1 and β1 subunits and is expressed in most cell and tissue types; however, two other sGC subunits, α2 and β2, have been identified, and mixed heterodimers have been discovered, in particular α2β1, which is expressed in the brain, placenta, spleen, and uterus.
Both α and β subunits contain the cyclase homology domain and belong to the class III cyclase family. Since the active site of sGC is located at the heterodimer boundary, two active centers are possible, but when subunits are connected, catalytic and pseudosymmetric pockets are formed [55].
Various physiological studies have shown that the β1 subunit of the heterodimer is necessary but not sufficient for proper sGC signaling; both α1 and α2 subunits can heterodimerize with the β1 subunit to form a functional protein. The conservative histidine on the β1 subunit was found to coordinate the heme moiety, which preferentially binds NO and carbon monoxide (CO) rather than oxygen (O2). Structural changes caused by the NO binding, activate the catalytic domain of the sGC protein, which causes vasorelaxation [56].
Elucidation of the NOS role in physiological and pathophysiological processes in the body NOS uncoupling as a basis for pathophysiological conditions
Under physiological conditions, eNOS produces NO, which is a key vasoprotective factor in the endothelium. However, under pathological conditions associated with an oxidative stress, eNOS may cease to function. The oxidative stress contributes to endothelial dysfunction primarily due to the rapid inactivation of NO via oxidation by excess superoxide. At the second stage, a constant oxidative stress renders eNOS unbound, whereby superoxide is produced at the expense of NO. The deficiency of the eNOS cofactor tetrahydrobiopterin (BH4), the deficiency of the eNOS substrate, L-arginine, and S-glutathionylation of eNOS are likely the main causes of the eNOS uncoupling. Peroxynitrite and superoxide can oxidize BH4 leading to the BH4 deficiency. The production of ROS from unbound eNOS has been shown in mouse models of atherosclerosis, as well as in patients with hypercholesterolemia, atherosclerosis, hypertension, diabetes mellitus, and in people who use nicotine for a long time [57].
Under conditions of atherosclerosis and vascular diseases, the bioavailability of NO in the vasculature decreases. All the known risk factors for cardiovascular diseases are accompanied by a decrease in NO bioavailability as a result of a decrease in its production and an increase in the superoxide inactivation.
Thus, a decrease in the NO production is not the result of a decrease in the eNOS expression. On the contrary, the eNOS expression is compensatory increased in vascular diseases. However, this compensation mechanism is often useless because the eNOS activity is inhibited or the eNOS enzyme becomes uncoupled (inoperative) [58].
Molecular mechanisms of NOS uncoupling
The most significant cause for NOS uncoupling is the loss of the critical cofactor BH4. It can be synthesized in two ways: in a three-step process from guanazine triphosphate (GTP) to BH4 and the so-called “rescue route”, which recycles oxidized 7,8 dihydrobiopterin back to BH4 using the recycling enzyme dihydrofolate reductase (DHFR, dihydrofolate reductase). An oxidative stress plays a significant role in the depletion of cellular BH4 pools, leading to uncoupling. Superoxide can directly oxidize BH4 to dihydrobiopterin (BH2) and, therefore, destabilize the NOS dimer, leading to uncoupling. This appears to be the main reason for NOS uncoupling during hypertension and ischemic reperfusion, as increased superoxide levels correspond to a decrease in the BH4/BH2 ratio and subsequent NOS uncoupling. The oxidative stress can also affect the bioavailability of BH4 not only through oxidation, but also through a direct decrease in the DHFR expression, thereby reducing the activity of the “rescue pathway” [59].
A direct oxidation of BH4 by superoxide is relatively slow, but BH4 is rapidly oxidized to BH2 by peroxynitrite, a highly reactive anion formed by the reaction of superoxide with NO. Peroxynitrite also causes significant cell damage by nitrating the thiol and hydroxyl groups of the amino acid side chain, thereby impairing the enzymatic function.
The consequences of NOS uncoupling include: reduced the de novo NO formation; binding of bioactive NO by superoxide anions through the formation of peroxynitrite; cellular damage mediated by superoxide and peroxynitrite; the peroxynitrite mediated oxidation of BH4 to BH2 leading to a further propagation of NOS uncoupling.
Thus, NOS uncoupling is a self-reinforcing biochemical process driven by BH4 oxidation. Due to the unbound eNOS-dependent oxidative stress, BH4 oxidation has been observed both in vitro and in vivo. In these experiments, uncoupling was induced by increasing the eNOS expression under the conditions of a stable expression of GTP-cyclohydralase (GTPCH-I), effectively reducing the ratio of BH4 to eNOS. If all other variables are held constant, the resulting oxidation of BH4 to BH2 can only be due to the formation of superoxide by the decoupled NOS. Cellular superoxide is also produced by a number of other mechanisms, including NADPH oxidase, xanthine oxidase, and redox processes. In pathology, the oxidative stress can induce BH4 oxidation, thereby initiating or maintaining NOS uncoupling. Excess superoxide resulting from the initial BH4 oxidation and NOS uncoupling will stimulate a cycle of cellular damage, a further BH4 oxidation (BH4 oxidation stabilizes unbound NOS subunits that continue to produce ROS and biochemical stabilization of the unbound NOS subunit activity). The interruption of this communication process is a promising strategy for the treatment of a wide range of cardiovascular diseases caused or exacerbated by the NOS uncoupling (Fig. 2) [60, 61].

Figure 2 – Synthesis, recycling and oxidation of BH4 as the determinant of NOS uncoupling. Note. On the left: Under normal conditions, BH4 bioavailability is maintained by de novo synthesis from guanosine triphosphate (GTP) in which the rate limiting step is catalyzed by GTP cyclohydrolase (GTPCH) by dihydrofolate reductase (DHFR) mediated recycling of 7,8-dihydrobiopterin (BH2), the primary product of a non-enzymatic oxidation of BH4. On the right: “Unbound” NOS are characterized by the formation of superoxide (O2-). NOS сleavage is facilitated by a decrease in the bioavailability of BH4 relative to the BH2 or NOS protein. In turn, O2- formed by unbound NOS, reacts with NO to form peroxynitrite (ONOO-), a highly reactive anion that rapidly oxidizes BH4. Hence, the NOS uncoupling state is stabilized by the self-propagating oxidative stress. In addition to this primary BH4-mediated cycle, additional mechanisms have been shown to promote uncoupling, including a reduced arginine bioavailability, high levels of oxidized glutathione (GSSG) compared to reduced glutathione (GSH), or elevated concentrations of endogenous NOS inhibitors LN-monomethylarginine (L-NMMA, monomethyl-L-arginine) and asymmetric dimethylarginine (ADMA). Adapted from [60].
Peroxynitrite leads to the NOS uncoupling. Although peroxynitrite can directly oxidize BH4 in vitro, it can also directly affect the enzyme itself, which also leads to uncoupling: peroxynitrite releases zinc from the zinc-thiolate cluster and destroys the NOS dimer (Fig. 3) [59].

Figure 3 – Pathological conditions and mechanisms of NOS uncoupling. Note. Hypercholesterolemia, hypertension, smoking, and diabetes mellitus lead to the activation of NADPH (a reduced form of nicotinamide adenine dinucleotide phosphate) oxidase, partially by a protein kinase C (PKC)-dependent mechanism. Diabetes mellitus also stimulates the production of mitochondrial ROS, that then triggers the activation of NADPH oxidase, which can increase a mitochondrial production of superoxide (O2-) and xanthine oxidase. The endothelial NO synthase (eNOS) enzyme can be uncoupled through two main mechanisms: deficiency of the BH4 cofactor or the L-arginine substrate. O2·- reacts with NO to form peroxynitrite (ONOO-). ONOO- oxidizes BH4 resulting in BH4 deficiency. L-arginine deficiency is caused by an increase in the arginase expression and activity, partially through RhoA/ROCK-dependent mechanisms. Unbound eNOS produces superoxide, thereby increasing the oxidative stress. The eNOS uncoupling reduces the endothelial NO production, which is further exacerbated by a decrease in the eNOS expression and activity. In addition to the population-level risk factors, the impaired blood flow makes arterial bifurcations and lateral branches prone to atherosclerosis. The increased oxidative stress and reduced endothelial NO production contribute significantly to the increased atherosclerosis in these areas. Adapted from [58].
S-glutathionylation is a reversible post-translational modification that adds glutathione to reduced cysteine residues. This most often occurs during the oxidative stress, as eNOS acts as one of the main targets for S-glutathionylation. It has been found out that the eNOS-dependent production of NO and superoxide is modulated by its S-glutathionylation, indicating that intense S-glutathionylation correlates with a decrease in the NOS activity and an increase in the superoxide production. The increased oxidative stress causes a proportional increase in the amount of S-glutathionylated eNOS, which also indicates that the production of superoxide by the upregulation principle is a trigger for the eNOS uncoupling [59].
Thus, S-glutathionylation uncouples eNOS, switching it from NO to the ROS formation, by attaching glutathione to cysteine residues between the FMN-binding and FAD-binding domains: this leads to enzyme uncoupling and the production of O2- free radicals. The oxidative stress triggers S-glutathionylation of eNOS in endothelial cells and intact vessels. In addition, S-glutathionylation is increased during vasoconstriction, which reduces endothelium-dependent vasodilation. Given the importance of NO and eNOS mediated endothelial dysfunction in diseases such as heart attack, stroke, diabetes and cancer, the identification of this new redox signaling pathway provides new insights into therapeutic approaches to prevent or treat them [62].
Drugs for prevention of NOS uncoupling
As it has been noted above, NOS uncoupling is one of the main factors in the development of various diseases and, therefore, a promising target for the pharmacological intervention. Many approaches have been taken to try restoring NOS in various diseases, including: blocking the upstream of the ROS production, the BH4 administration, the administration of folic acid to recycle BH2 to BH4, and the L-arginine administration (Fig. 4).

Figure 4 – Schematic representation of known causes and methods for correcting NOS uncoupling in the cardiovascular system. Note. Causes for NOS uncoupling are shown in red and treatments are shown in blue. Superoxide (O2-) or peroxynitrite (ONOO-) can oxidize BH4 to BH2, which can be prevented by antioxidants or NADPH oxidase inhibitors, and interfere with s-glutathionylation (s-Glu). Folic acid can stimulate dihydrofolate reductase (DHFR) to reduce BH2 to BH4. Arginase inhibition can preserve L-arginine levels to make it available to the NOS enzime. Adapted from [59].
BH4
A decreased BH4 bioavailability followed by the eNOS uncoupling has a significant impact on the pathogenesis of endothelial dysfunction, which is a hallmark of vascular damage in cardiovascular pathologies, including hypertension, hyperlipidemia, and diabetes [63]. The intracellular ratios of BH4/eNOS (and of BH4 and other NOS isoforms) remain poorly defined, making it unclear whether the intracellular BH4 deficiency is sufficient to induce the eNOS uncoupling. The BH4/BH2 ratio and the absolute molar concentration of BH4 are key determinants of eNOS binding in vivo. The degree of BH4 oxidation, BH2 accumulation, and the superoxide production are directly correlated with the intracellular eNOS/BH4 ratio. Through the mechanisms independent of eNOS, such as a direct uptake of reactive oxygen species, BH4 can have a significant impact on the levels of the reactive oxygen species production by cells [64].
Thus, the replenishment of intracellular BH4 can be considered a promising approach to stabilize NOS. However, there are reasons that limit the clinical application of this approach. The oral administration of excess BH4 may be not effective if the NOS isoforms ratio is not balanced: too much BH4 can activate iNOS under the conditions of inflammation, exacerbating the pathophysiology of this process. BH4 administered p. o., is likely to oxidize and needs to be regenerated, but the attempts to enhance this re-regeneration process by combining BH4 with antioxidants (vitamin C) have not resulted in the improved clinical efficacy of this combination. BH4 has already been approved by the Food and Drug Administration (FDA, https://www.fda.gov/) as a therapeutic agent for the treatment of phenylketonuria, a phenylalanine hydroxylase deficiency that can be partially corrected by the administration of BH4. Sapropterin dihydrochloride (Kuvan®, BioMarin, Tiburon, CA, www.vidal.ru) is a synthetic version of BH4 currently used to treat patients with phenylketonuria [65, 66].
An analog of BH4 is the compound 6-acetyl-7,7-dimethyl-5,6,7,8-tetrahydropterin (ADDP), a stable compound, soluble in both polar and organic solvents. ADDP can diffuse from plasma through cell membranes and cause vasodilation by stimulating the eNOS activity [67].
Metformin can attenuate the oxidative damage to endothelial cells by increasing the levels of GTP cyclohydralase-I (GTPCH-I) and BH4, and thereby preventing the eNOS uncoupling. Metformin also inhibits the p47-phox subunit of NADPH oxidase, thereby reducing the ROS production and BH4 oxidation. These protective effects of metformin are partially mediated by the activation of the adenosine monophosphate-activated protein kinase (AMPK) signaling pathway [68].
Arginase inhibitors
The role of the arginase overexpression and/or activity and its downstream targets in cardiovascular dysfunction and injury, has been well established. Pharmacological effects on specific components of the arginase/ornithine pathway are promising as therapy for cardiovascular diseases [69].
Ureohydrolase arginase is a manganese-containing enzyme that catalyzes the final step of the ornithine cycle by converting L-arginine to L-ornithine and urea. The two isoforms of this enzyme have similar mechanisms of action and produce the same metabolites. The homology of their amino acid residues is more than 60%, and the regions important for the function of the enzyme are 100% homologous. In addition, both isoforms are involved in the dysregulation of the NOS function [70].
The arginase-induced uncoupling of eNOS leads to the increased ROS production, which contributes to the endothelial dysfunction and increased vascular stiffness. Increasing the level of arginase can reduce the concentration of L-arginine and has been shown to contribute to the pathophysiology of the circulatory system diseases [71].
Preclinical studies have shown that the addition of L-arginine can improve the erectile dysfunction, prevent or reduce the severity of the endothelial dysfunction. However, some studies on animals and humans have found out no benefit or increased adverse effects from the chronic administration of L-arginine. The resulting side effects are likely related to the counter-regulatory effects of L-arginine in the increasing arginase expression/activity, which may reduce the availability of L-arginine for NOS. Under the conditions of the excessive arginase activity, a competition with NOS for L-arginine causing the enzyme uncoupling, will occur. The increased formation of reactive oxygen species and key inflammatory mediators contributes to a pathological increase in the arginase activity [71].
Thus, several amino acids inhibit arginase, including: L-ornithine, L-leucine, L-valine, L-lysine, L-isoleucine, and L-norvaline. L-ornithine is the most potent of the above-listed. In addition, L-citrulline increases the NO production, but is also an allosteric inhibitor of arginase [71].
The research by El Bassossy H.M. et al. showed that the inhibition of arginase by citrulline, norvaline, or ornithine, alleviates hypertension associated with a metabolic syndrome by direct and indirect defense mechanisms. The direct mechanism is to maintain an endothelium-dependent relaxation and the NO generation, while the indirect mechanism is realized through the inhibition of insulin resistance and hypertriglyceridemia [72]. Ornithine inhibits the enzyme arginase by competing with its substrate (L-arginine) at the active sites of the enzyme. L-norvaline is a noncompetitive inhibitor of the enzyme arginase, and L-citrulline is its allosteric inhibitor.
L-norvaline (or 2-aminopentanoic acid) is a non-proteinogenic amino acid and an isoform of the common amino acid valine that has been extensively investigated in early enzymological studies. The structural similarity with L-ornithine gives the substance the ability to inhibit arginase by the negative feedback principle. In addition, the anti-inflammatory properties of L-norvaline have been demonstrated in vitro through the inhibition mechanism of ribosomal protein kinase S6-kinase β1. It should be notified that the arginase inhibition is promising for reducing the risk and frequency of cardiovascular diseases [73].
Statins inhibit HMG-CoA reductase, which catalyzes the conversion of HMG-CoA to mevalonic acid, limiting cholesterol biosynthesis in the liver. In addition to inhibiting cholesterol synthesis, statins also block the synthesis of intermediate isoprenoids such as farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP). FPP and GGPP serve as important lipid components for post-translational modification of various proteins, including heterotrimeric G proteins and small GTP-binding proteins belonging to the Ras, Rho, Rap, and Rab family of GTPases. Isoprenylation is critical for the intracellular transport and the function of small GTP-binding proteins. The modification with FPP is required for a proper localization of Ras family proteins, while GGPP is required for Rho, Rab and Rap family proteins. However, some Rho GTPases require both farnesylation and geranylgeranylation for proper functioning and the intracellular localization. By inhibiting the synthesis of mevalonate, statins inhibit the synthesis of intermediate isoprenoids, thereby preventing the isoprenylation of small GTPases, which leads to the inhibition of these signaling molecules. Interestingly, some of the cholesterol-independent or “pleiotropic” effects of statins may be related to the ability of statins to block the synthesis of intermediate isoprenoids [74, 75].
The ability of statins to increase the eNOS expression and activation may be an important mechanism by which statins improve the endothelial function and lower cholesterol levels (Fig. 5).

Figure 5 – Effect of statins on eNOS in endothelial cells. Note. By inhibiting mevalonate synthesis, statins reduce GGPP levels, prevent the Rho/ROCK activation and stabilize eNOS mRNA, thereby increasing the eNOS expression. By reducing caveolin-1 and circulating ADMA, as well as increasing eNOS phosphorylation at the Ser-633 and Ser-1177 activation sites via AMPK, Akt, and PKA, statins also increase the eNOS activity. Finally, by upregulating GTPCH, statins increase endothelial BH4, the presence and restoration of eNOS binding. GGPP is geranylgeranyl pyrophosphate; ROCK – Rho-associated protein kinase; GTPCH – guanosine triphosphate cyclohydrolase; AMPK 2 adenosine monophosphate activated protein kinase; PKA, protein kinase A; PI3K – phosphatidylinositide-3-kinase; ADMA - asymmetric dimethylarginine. Adapted from [77].
Geranylgeranylation of Rho GTPase has been shown to result in the downregulation of eNOS in endothelial cells; the administration of statins inhibited the formation of GGPP and therefore resulted in an increase in the eNOS expression, which was not observed during the co-incubation with GGPP. It has also been shown that an increased level of endogenous low-density lipoproteins (LDLs) negatively affects the expression of eNOS mRNA in human umbilical vein endothelial cells (HUVECs), and this effect, at the post-transcriptional level, was neutralized after the administration of simvastatin. The mechanism by which statins increase the eNOS mRNA stability is through an increased polyadenylation of eNOS mRNA due to the changes in the actin cytoskeleton caused by the Rho inhibition. In addition, the statin treatment of HUVECs induced by hydrogen peroxide (H2O2) activates the phosphatidylinositide 3-kinase (PI3K, phosphoinositide 3-kinase)/Akt pathway and enhances the eNOS expression. Tumor necrosis factor alpha (TNFα) is a powerful inflammatory stimulus that activates miR-155 miRNA and, accordingly, eNOS mRNA. It is important to notify, that its (TNFα) activation was attenuated by simvastatin, but the positive effect of the statin was abolished by co-incubation with mevalonate or GGPP [76].
In addition to increasing the eNOS expression at the transcriptional and post-transcriptional levels, at the post-translational level, statins enhance the activity of eNOS, which has multiple phosphorylation sites by serine/threonine residues. A HUVEC exposure to fluvastatin increases eNOS phosphorylation at the Ser-633 and Ser-1177 activation sites via protein kinase A (PKA) and the PI3K/Akt-mediated pathway, respectively. The statin-mediated induction of eNOS phosphorylation at Ser-1177 depends on the recruitment of Akt to the eNOS complex in endothelial cells (heat shock protein-90). In endothelial cells, atorvastatin increases eNOS phosphorylation at Ser-633 via adenosine monophosphate-activated protein kinase (AMPK). Moreover, the ex vivo incubation of rat mesenteric resistant arteries with simvastatin, induces rapid AMPK-mediated eNOS phosphorylation at Ser-1177. In addition to modulating phosphorylation, statins also increase the eNOS activity at the post-translational level, causing it to dissociate from caveolin-1. Atorvastatin reduces the content of caveolin-1 and activates eNOS in EC, regardless of the presence or absence of LDL cholesterol [77].
Statins act at several levels, increasing both eNOS activity and stabilizing its dimers. The exposure of HUVEC to cerivastatin or fluvastatin results in the increased GTPCH expression and the increased BH4 bioavailability, resulting in improved eNOS binding. In addition, statins prevent the oxidation of tetrahydrobiopterin to trihydrobiopterin (BH4 to BH3). The result of these processes is a decrease in the oxidative stress in endothelial cells and an improvement in the eNOS function [76].
Asymmetric dimethylarginine (ADMA) acts as an endogenous inhibitor of eNOS. ADMA is produced as a by-product of protein metabolism due to methylation of L-arginine residues, its production is significantly increased by LDL in endothelial cells, and it is metabolized by the enzyme dimethylarginine dimethylaminohydrolase (DDAH); under physiological conditions, it is excreted in the urine. Due to its close structural similarity to L-arginine, ADMA suppresses the eNOS activity either through the inhibition of the L-arginine cellular uptake or through a direct competitive inhibition of eNOS binding [78].
There are several hypotheses about the ways statins affect ADMA levels. One of them concerns the inhibition of the ADMA-induced inflammatory response modulated by mitogen-activated protein kinase (MAPK) in human endothelial cells. Statins activate the transcription factor of sterol response element binding protein (SREBP) by lowering membrane cholesterol. This factor specifically enhances the expression of more than 30 genes associated with the synthesis and absorption of fatty acids, phospholipids, cholesterol and triglycerides. One of its isoforms, a sterol regulatory element binding protein 2 (SREBP-2), increases the transcription of proprotein convertase subtilisin/kexin type 9 (PCSK9, proprotein convertase subtilisin/kexin type 9). Statins increase both PCSK9 mRNA and LDLR levels by activating sterol-mediated SREBP-2, an important activator of the transcription and DDAH activity (Fig. 6) [79].

Figure 6 – List of the most important inhibitors based on arginine. Note. Adapted from [92].
Resveratrol
Resveratrol (3,5,4’-trihydroxy-trans-stilbene) is a phytoalexin polyphenol found in various plant species including Veratrum grandiflorum (Maxim. ex Baker) O. Loes, Polygonum cuspidatum Siebold & Zucc., Vitis vinifera L., Arachis hypogaea L. and Morus rubra L. The name “resveratrol” comes from its origin; the compound is a resorcinol derivative from the species of the genus Veratrum.
Molecular targets of resveratrol include those which interact directly with it and the others that are modulated indirectly (e.g., through changes at the expression levels). For its effect on eNOS, the following targets are of particular importance: NAD+ – dependent histone deacetylase sirtuin 1 class III (SIRT1, silent information regulator 1); AMP-activated protein kinase (AMPK); nuclear factor-derived erythroid 2 – dependent factor-2 (Nrf2, nuclear factor E2-related factor 2); estrogen receptors (ER).
A part of the resveratrol effects on the cardiovascular system is mediated by endothelial NO. Resveratrol enhances the NO production through several mechanisms and prevents the NO breakdown by reducing the oxidative stress (Fig. 7).
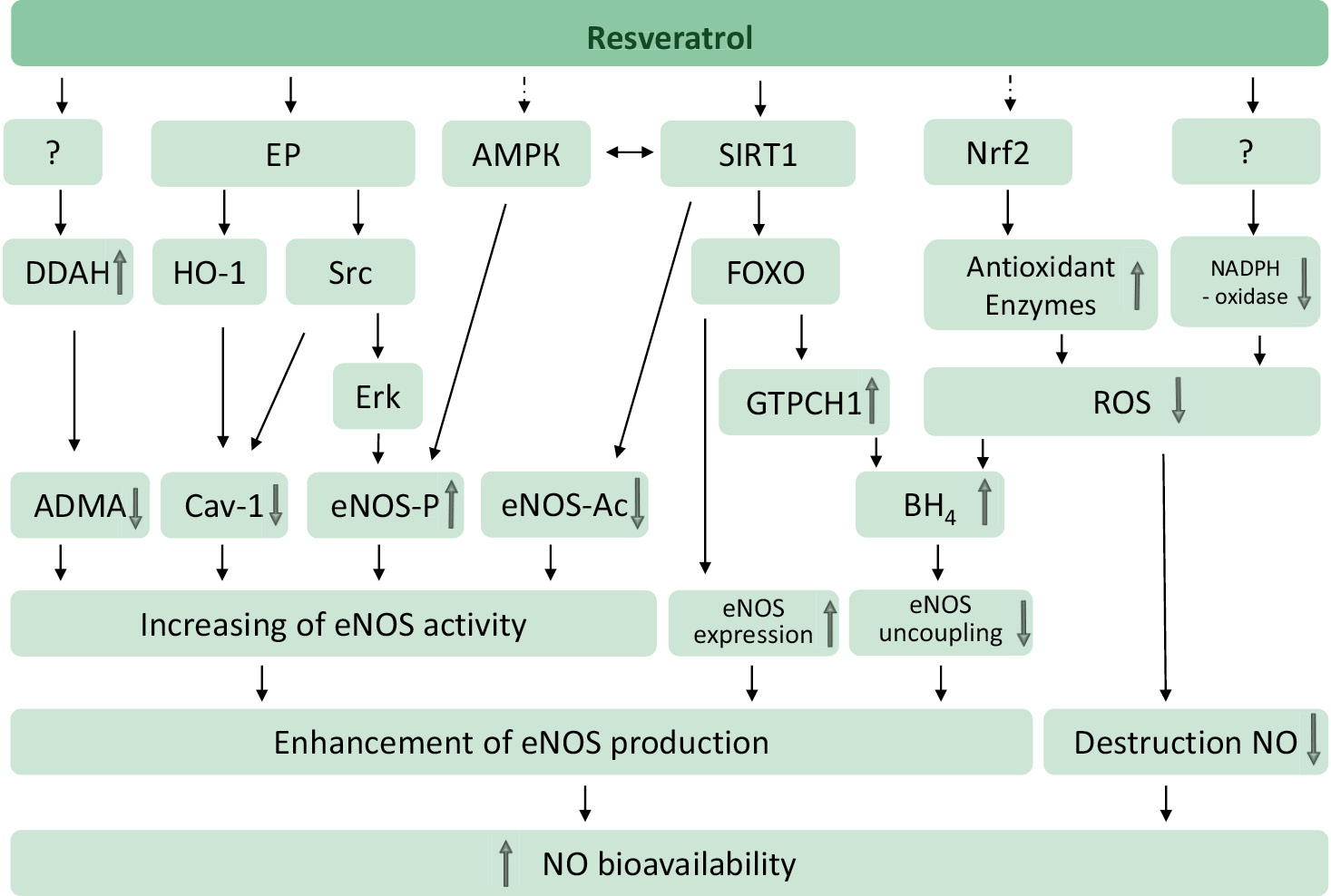
Figure 7 – Resveratrol targets to prevent NOS uncoupling (80 adapted). Note. Resveratrol increases the NO production and prevents its breakdown. Resveratrol can activate sirtuin 1 (SIRT1) either directly (in a substrate-dependent manner) or indirectly (by inhibiting phosphodiesterases). SIRT1 stimulates endothelial NOS via deacetylation, enhances the eNOS expression via deacetylation of the forkhead box protein O1 (FOXO) transcription factor, and prevents the eNOS uncoupling by increasing the activity of GTP-cyclohydrolase I (GTPCH-I), a limiting factor in BH4 biosynthesis. AMPK and nuclear factor 2 associated with Nrf2 are indirect targets of resveratrol. AMPK phosphorylates eNOS by serine 1177. eNOS can also be phosphorylated by Erk1/2, which is stimulated by a pathway involving the estrogen receptor (ER) and the Src family tyrosine kinase. Caveolin-1 (Cav-1) is a protein that negatively regulates the eNOS activity. ADMA is an endogenous eNOS inhibitor that is cleaved by DDAH. The targets for the activation of dimethylarginine dimethylaminohydrolase (DDAH) or inhibition of NADPH oxidase have not been identified yet. Adapted from [80].
Activators of various NOS isoforms
Calcium dobesilate (Doxium 1)
Calcium dobesilate (CaD) is widely used as an angioprotective agent for the treatment of vascular diseases, inhibits platelet aggregation, thrombus formation, increased capillary permeability, reduces their fragility, increased blood viscosity.
A specific property of dobesilate is a moderate increase in the eNOS activity with little or no effect on the iNOS activity, which indicates its future outlook for the treatment of vascular diseases, especially those caused by impaired NO secretion in the endothelium, including prolonged hyperglycemia [81].
The mechanism of calcium dobesilate action on the eNOS expression is realized in several ways.
As a result of the study of the molecular and cellular mechanisms underlying the protective effects of CaD carrird out by Zhou Y. et al. [84], the effect of calcium dobesilate on pentraxin 3 (PTX3) was established. This protein is an important component of humoral immunity and is secreted by various cells, including endothelial, mononuclear macrophages, fibroblasts, adipocytes, dendritic and smooth muscles after the stimulation of the inflammatory response. High plasma levels of PTX3 are associated with many diseases conjugated with endothelial dysfunction (diabetes mellitus, arterial hypertension, chronic kidney disease). PTX3 interferes with eNOS phosphorylation by serine 1177, reducing NO production, leading to endothelial dysfunction.
PTX3 induces dysfunction and morphological changes in the endothelial layer via the P-selectin/matrix metalloproteinase-1 pathway, which prevents the separation of eNOS from caveolin-1, resulting in impaired NO signaling [82]. Another study also confirms that PTX3 inhibits the NOS/NO pathway by inhibiting eNOS phosphorylation and therefore not inducing a detachment from caveolin-1 required for activation [83].
Thus, calcium dobesilate can inhibit the PTX3 expression by altering the IKK/IKB/NF-κ B pathway, thereby improving endothelial dysfunction at the cellular level [84].
Endothelial cells also need a vascular endothelial growth factor A (VEGF-A) to maintain their various functions (differentiation, permeability, and the NOS expression) and survive [85], however, an excessive amount of this factor causes their damage, increased glomerular permeability and inflammation. An appropriate balance of the VEGF expression, binding, and signaling is required to maintain glomerular health and prevent glomerulosclerosis and rarefaction. Under pathological conditions, this balance is disturbed [86].
Despite high concentrations of VEGF in the vasculature, a reduced NO response was called “uncoupling” of eNOS and VEGF, which leads to the disruption of the cellular response and the intensification of the pathological process course. In addition to its positive effect on eNOS, at the molecular level, calcium dobesilate also reduces the oxidative stress and inhibits growth factors such as fibroblasts growth factor (FGF) and VEGF [87].
CavNOxin
CavNOxin is a synthetic peptide of 20 amino acids in the framework domain of caveolin with a triple alanine substitution fused to the peptide penetratin for a free entry into cells.
CavNOxin increases NO levels, decreases a vascular tone, and lowers mean BP in wild-type mice, but the effect is lost in gene or caveolin-1 knockout animals, indicating that it is caveolin-1 specific.
A chronic CavNOxin administration reduces the severity of atherosclerosis in mice with apolipoprotein E (ApoE, apolipoprotein E) knockout, fed a high-fat diet, and in mice with diabetes and atherosclerosis, while the loss of eNOS genetically reduces its effect. These results suggest that CavNOxin may reduce the inhibitory effect of caveolin on the eNOS function, allowing increased endogenous NO biosynthesis. The data from genetic studies using transgenic mice with the endothelial-specific expression of F92A CAV1 confirm that the regulated expression of modified caveolin is sufficient to increase the NO bioavailability, which leads to a significant decrease in BP [88].
NOS transcription enhancers
AVE3085 and AVE9488 are two small molecules that have been identified by high-throughput screening able of enhancing the eNOS67 transcription in vitro and in vivo.
A promoter analysis using the luciferase test showed that both compounds bind even the shortest fragment of the eNOS promoter (263 polynucleotides), but do not overlap with known eNOS transcription factors (GATA, Sp1/3-like, Elf-1, YY1, Sp1 or PEA3).
AVE3085 has a promoter binding cis element located in the proximal 263 base pairs of the promoter region.
In the experiments on the human internal mammary artery obtained after a coronary artery bypass surgery, a positive effect of AVE3085 was shown. It was expressed in the prevention of the damaging effect of homocysteine on the endothelium during the co-incubation with AVE3085, while an increase in the level of eNOS mRNA and an increase in the NO production was observed [89].
These NOS transcription enhancers have also been shown to increase vascular BH4 levels, reduce a cuff-induced neointima formation, and improve the course of atherosclerosis in ApoE knockout animals after 12 weeks of diet. AVE9488 promotes cardiomyocyte remodeling in a model of myocardial infarction, which can be explained by a decrease in SMAD-mediated signaling [90].
Blockers of various NOS isoforms NOS inhibitors can be classified from different points of view.
A classical enzymological approach makes it possible to distinguish reversible (subgroups: competitive, non-competitive and mixed type), irreversible, as well as reactive inhibitors, the action of which depends on the enzymatic reaction.
The most widely used classification of inhibitors is based on the site of their binding to the NOS enzyme, which makes it possible to recognize four different classes of inhibitors.
The first class, in fact the largest, interacts with the arginine-binding site. Some of the compounds belonging to this class are reaction-level inhibitors because they require an active enzyme and NADPH for a complete inhibition. The second class includes a set of compounds that mimic the BH4 cofactor. The third class is represented by inhibitors that directly interact with heme. Some inhibitors belonging to this class bind to the heme of the monomeric form of the enzyme and prevent the formation of its active dimer. The fourth class covers NOS inhibitors interacting with calmodulin or flavin cofactors [91].
There is also a classification according to which NOS inhibitors can be divided into two groups: based on amino acids (derivatives and close analogues of arginine) and based on non-amino acids (a wide range of compounds that do not have an arginine-like amino acid backbone). Arginine-based NOS inhibitors have generated a particular interest as this category includes a large number of compounds with potential experimental and clinical applications. Since these compounds are readily available and stable in the aqueous media, they serve as an excellent tool for an experimental inhibition of NOS [91].
Non-selective inhibitors
Even before crystal structures had been created, arginine-based compounds were found to inhibit NOS and exhibit neuroprotective properties. For example, NG-mono-methyl-L-arginine (2 L-NMMA) and NG-nitro-L-arginine methyl ester (3 L-NAME) protect brain neurons in a stroke and Parkinson’s disease. However, these inhibitors also cause hypertensive effects, most likely due to the inhibition of eNOS.
The earlier studies have shown that L-arginine substrate analogs have inhibitory properties against three NOS isoforms. Among them, NG-monomethyl-L-arginine (L-NMMA) and NG-nitro-L-arginine (L-NNA) and its precursor NG-nitro-L-arginine methyl ester (L-NAME) have been characterized as common NOS inhibitors and continue to be widely used in research, especially L-NAME [92].
Endogenous ADMA and L-NMMA inhibitors
There are two endogenous NOS inhibitors. ADMA is a potent, non-competitive NOS inhibitor, while its affined L-NMMA is a less potent, competitive NOS inhibitor. Although ADMA has been shown to contribute to the development of the inflammatory syndrome and endothelial dysfunction observed in shock, the possibility of its clinical application requires a further study [91].
546C88 (N(G)-methyl-L-arginine hydrochloride)
Compound 546C88 (N(G)-methyl-L-arginine hydrochloride) is a non-selective NOS inhibitor that appears to rebalance a vasomotor tone in patients with septic shock, by reducing the concomitant need for norepinephrine. It has been studied in phase III clinical trials in Europe, North America, South America, South Africa and Australia. This study was terminated in advance due to an increased risk of death [91].
L-NAME
N-nitro-L-arginine methyl ester (L-NAME) and Ng-nitro-L-arginine (L-NArg) are synthetic, non-selective NOS inhibitors with implications for a substance abuse because they attenuate signs of opioid withdrawal in rats. L-NAME also seems promising for the treatment of a septic shock by maintaining adequate BP levels. A chronic administration of L-NAME reduces angiogenesis during in vitro migration and invasiveness, pointing to its possible future use as a tumor growth inhibitor [91].
VAS203
Pterin 4-amino-tetrahydrobiopterin (VAS203) is a BH4 analogue that can inhibit all NOS proteins by replacing the BH4 cofactor. VAS203 has shown efficacy in the treatment of the traumatic brain injury and has been used in phase II clinical trials [92].
Selective inhibitors of iNOS
N-[3-(aminomethyl)benzyl]acetamidine
N-[3-(aminomethyl)benzyl]acetamidine is a specific iNOS inhibitor. It remains one of the most widely used NOS inhibitors in the research due to its cell and tissue permeability. Its selectivity appears to be associated with an irreversible effect on the more rapidly responsive iNOS, while the inhibition of nNOS and eNOS is reversible. The similar effect is observed in N(5)-(1-iminoethyl)-1-ornithine (1-NIO) [92].
7-nitroindazole
One of the first highly selective compounds is 7-nitroindazole (7-NI). Initial crystallographic studies showed that 7-NI binds at the active site of eNOS and changes the Glu orientation of the active site, while 3-bromo-7-NI can bind at both the active and pterin sites [93]. 7-nitroindazole is a specific nNOS inhibitor and exhibits an anticonvulsant effect on models of seizures in rodents. It has also been shown to occasionally induce convulsions in rodents caused by cainite, nicotine, and soman. Although it has been shown that all three isoforms can bind 7-NI with very similar affinity, in vivo studies have shown inhibition of nNOS without significant effect on BP. This may be due to the fact that 7-NI can be taken up by cells in different ways and the permeability of endothelial cells for this compound is limited. Thus, although 7-NI cannot be accurately described as a specific inhibitor of nNOS, it may behave as such in in vivo models [92].
Aminoguanidine
Aminoguanidine is a selective iNOS inhibitor that attenuates the “graft versus host” disease by reducing hematopoietic parameters and a concomitant susceptibility to the bacterial infection in mice [94].
L-NIL
An exceptional compound among the first arginine-based NOS inhibitors developed is L-NIL. It has a moderate selectivity for iNOS, is well tolerated, and its prodrug (L-NIL-TA) can be administered orally [95].
GW273629 и GW274150
Compounds GW273629 and GW274150 are sulphur-substituted acetamidine amino acids that are specific iNOS inhibitors. Having safer toxicity profiles than 1400W, GW273629 and GW274150 have been used in clinical trials for migraine therapy (NCT00242866; NCT00319137). The both compounds were found to be ineffective, possibly due to the peculiarities of pharmacokinetics [91, 92, 95].
eNOS
As mentioned above (see section “Endothelial NO synthase”), eNOS is posttranslationally palmitylated and transported to caveolae, where its activity is reduced due to binding to caveolin-1. Based on this mechanism, it seems possible to mimic the function of caveolin-1.
Based on this approach, cavtratin, a drug that inhibits eNOS, was developed. It is a caveolin scaffold domain fused to the cell-permeable peptide penetratin, which allows caveolin to pass through the plasma membrane. Cavtratin significantly targets at eNOS, reducing its activity and mimicking the inhibitory effect of caveolin-1, while not affecting the function of other NOS in vivo. Cavtratin reduces an increased microvascular permeability and blocks a tumor progression (shown in the model of HepG2 hepatocarcinoma cells transplanted into mice without thymus), restores tight junctions in brain microvascular endothelial cells treated with chemokine-2, inhibits matrix metalloproteinase-2/9 and cyclooxygenase-2, which can lead to the stabilization of atherosclerotic plaques and reduce the risk of intracerebral hemorrhage. Cavtratin also reduces the growth of glial cells and has a protective effect on the blood-brain barrier when exposed to VEGF-A in a mouse model of multiple sclerosis.
It remains unknown whether eNOS is the only target of cavtratin. However, most of the blocking effects of VEGF and the Transforming growth factor (TGF) were reversed when the experiments were performed in eNOS-deficient mice.
It is possible that cavtratin may increase blood pressure during a course administration. However, there is no evidence to support this potential effect. Furthermore, since CAV1 is believed to regulate many intracellular signaling pathways, cavtratin may have pleiotropic effects on many tissues, especially those lacking eNOS [90].
Other approaches to inhibition
Inhibition of downstream mediators
Methylene blue is a soluble guanylate cyclase (sGC) inhibitor. It is a dye that can be safely used in the people with a septic shock. It provides an effective protection in the TNFα-induced shock as well as in anaphylactic hypotension not corrected by other pharmacological agents. Methylene blue may be also useful in refractory cases of vasoplegia, a common complication of cardiopulmonary bypass, by alleviating an inflammation-mediated dysregulation of the endothelial function [96]. 1H-[1, 2, 4]oxadiazolo-[4, 3-a]quinoxalin-1-one (ODQ) is a selective sGC inhibitor in rats that restores a basal ganglia function and improves motor symptoms in Parkinson’s disease [94].
NO recycling
An NO-sensitive hydrogel, which demonstrated its fast response and sensitivity to NO, was developed by the research group of Park J. et al. That presented a potential for the application in biology and medicine [96]. There are projects to evaluate the ability of NO-cleavable cross-linkers (NOCCLs) to capture NO for the treatment of rheumatoid arthritis (RA). This is the first study on the possibility of using endogenous NO directly for the treatment of RA. By controlling the concentration of acrylamide as a polymer base and NOCCL as a crosslinking agent, a spherical nanogel was prepared by a solution polymerization to obtain an NO-neutralizing nanosized hydrogel (NO-Scv gel) for treating rheumatoid arthritis. As expected, the NO-Scv gel took up NO and reduced the levels of pro-inflammatory cytokines produced by activated macrophages in vitro. In addition, after an intra-articular injection of the nanogel into each paw of the mice with experimental RA, the therapeutic effects observed were superior to those notified with in vivo dexamethasone [98], which inhibits an iNOS-dependent NO formation by destabilizing iNOS mRNA by a mechanism requiring de novo protein synthesis. The effect of dexamethasone on the iNOS expression and the NO production, as well as on the degradation of iNOS mRNA, is to some extent abolished by type II interferon [97].
Therapeutic applications of activity modulators of various NO synthase isoforms
After various NOS isoforms had been identified, the search for their inhibitors progressed rapidly. As more and more active molecules were becoming available, it has become clear that the selectivity for NOS isoforms is a major problem. All 3 isoforms of human NOS (nNOS, eNOS, and iNOS) have nearly identical active site structures, making it difficult to design a selective inhibitor. It is especially important to avoid inhibition of eNOS due to its vital role in the cardiovascular system.
Experimental approaches using modulators of nitric oxide synthesis, including substrate manipulations, NO synthesis donors and inhibitors, will be useful to establish the relationship between NO systems and the cells of the cardiovascular and renal systems.
As a vasodilator, NO may be a unique therapeutic agent for the treatment of hypertension and, as a result, renal failure and left ventricular hypertrophy. The inclusion of NO modulators in clinical practice may be useful not only as therapeutic agents for certain diseases, but also as means to slow the progression of diseases due to their influence on many systems of the mammalian body.
As a therapeutic approach, a modulation of the NOS activity can be implemented to treat endothelial dysfunction, which is the cause for many diseases. For example, the development of endothelial dysfunction is characteristic of diabetes mellitus: the course of this disease is characterized by a number of metabolic disorders, which ultimately lead to the disruption of functioning of various organs and systems, mainly cardiovascular. A summary of the characteristics of various NOS is presented in Table 1.
Table 1 – Characteristics of various NO-synthase isoforms
Names | NOS-1 nNOS Neutral NOS NOSI | NOS-2 iNOS Induced NOS NOSII | NOS-3 eNOS Endothelial NOS NOSIII |
Monomer size (person) | ~160 кDа | ~131 кDа | ~133 кDа |
Cofactors | NADPH; FMN; FAD; BH4; Calmodulin; heme | ||
Inhibitors | N-methyl-L-arginine N-nitro-L-arginine 7-nitroindazole 1-(2-trifluoromethylphenyl)imidazole L-thiocitrulline S-methyl-L-thiocitrulline | Aminoguanidine S-benzylisothiourea 1-(2-trifluoromethylphenyl)imidazole L-N6-(1-iminoethyl)lysine 1400W | N-methyl-L-arginine N-nitro-L-arginine N-iminoethyl-L-ornithine 7-nitroindazole |
Intracellular localization | Cytosol; Plasma membrane [99]; Mitochondria [100]; Caveoli [101]. | Plasma membrane [102]; Phagosomes [103]; Golgi complex; Mitochondria. | Plasma membrane; Lipid rafts; Caveoli [104]; Cytosol [100]. |
Cells | Neurons [105]; Islets of the pancreas [106]; Endothelial cells; Smooth muscle cells [107]. | Macrophages and other leukocytes [108]; Smooth muscle cells; Cardiomyocytes [35]. | Endothelial cells [109]; Platelets [25]. |
Tissue expression | Central and peripheral nervous system [105]; Skeletal muscles [110]; Endometrium [111]; Macula densa [112]. | Heart; Liver [113]; Smooth muscles; Endothelium [113]. | Endothelium [114]. |
Functions | Neurotransmission [105]; Renal tubular glomerular interactions [115]; Intestinal peristalsis [116]. | Bactericidal activity; Cytotoxicity; Inflammation [117]; Septic shock [118]. | Vascular relaxation Decreased platelet adhesion [119, 120]; Angiogenesis [121]. |
Involvement in diseases | Renal diseases [115]; Affective disorders [122]. | Inflammation | Hypertension; Endothelial dysfunction. |
CONCLUSION
Enzymes of the NOS group became pharmacotherapeutic targets immediately after their key role in the NO biosynthesis had been established, and the interest in them is growing as new aspects of the pathogenesis of many acute and chronic diseases are being elucidated. Currently, almost all human diseases are to some extent associated with the endothelium and its dysfunction, and the restoration of the adequate vascular homeostasis is impossible without blood flow maintaining in the microvasculature at the physiological level. Increasingly, therapeutic strategies for many socially significant diseases (metabolic, cardiovascular, and neurometabolic) are endotheliocentric in nature and are aimed at preventing primary or secondary cardiovascular complications. Today, approaches to the stimulation of angiogenesis factors are being studied for the treatment of cardiovascular diseases, which show promising results in animals, but have not yet been implemented in the clinic. One important reason for this is the complexity and a large number of related molecular mechanisms that regulate ischemic vascular remodeling. Being responsible for the coordination and activity of vascular factors, the NO molecule can be such a regulator. Therefore, the NOS activity modulation may thus serve as an important factor for the operation of the complex network, contributing to a greater efficiency of angiogenesis.
FUNDING
The work was supported by the Russian Science Foundation (Project No. 20-75-10013).
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
AUTHORS’ CONTRIBUTION
Denis V. Kurkin, Elizaveta E. Abrosimova, Dmitry A. Bakulin, Evgeniy I. Morkovin, Ivan N. Tyurenkov – conception, article planning, review of literary sources, collection of materials, writing and editing the article; Nikolai S. Kovalev, Marina A. Dubrovina, Aleksandr V. Borisov, Andrey V. Strygin – collecting materials, editing the article.
About the authors
Denis V. Kurkin
Volgograd State Medical University
Author for correspondence.
Email: strannik986@mail.ru
ORCID iD: 0000-0002-1116-3425
Doctor of Sciences (Pharmacy), Associate Professor, Professor of the Department of Clinical Pharmacology and Intensive Care, First Deputy Director of the Research Center of Innovative Pharma Products
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Elizaveta E. Abrosimova
Volgograd State Medical University
Email: abrosimova.volgmed@gmail.com
ORCID iD: 0000-0002-6472-6906
post-graduate student of the Department of Pharmacology and Pharmacy, Institute of Continuing Medical and Pharmaceutical Education
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Dmitry A. Bakulin
Volgograd State Medical University
Email: mbfdoc@gmail.com
ORCID iD: 0000-0003-4694-3066
Candidate of Sciences (Medicine), Senior Researcher, Laboratory of Pharmacology of Cardiovascular Drugs, Research Center of Innovative Pharma Products
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Nikolai S. Kovalev
Volgograd State Medical University
Email: kovalev.volgmed@gmail.com
post-graduate student of the Department of Pharmacology and Pharmacy, Research Center of Innovative Pharma Products
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Marina A. Dubrovina
Volgograd State Medical University
Email: dubrovina.volgmed@gmail.com
ORCID iD: 0000-0003-1903-8589
post-graduate student of the Department of Pharmacology and Pharmacy, Research Center of Innovative Pharma Products
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Aleksandr V. Borisov
Volgograd State Medical University
Email: borissow1978@rambler.ru
ORCID iD: 0000-0003-0202-0008
Researcher, Laboratory of Pharmacology of Cardiovascular Drugs, Research Center of Innovative Pharma Products
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Andrey V. Strygin
Volgograd State Medical University
Email: drumsav@mail.ru
ORCID iD: 0000-0002-6997-1601
Candidate of Sciences (Medicine), Associate Professor, Deputy Director of the Research Center of Innovative Pharma Products
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Evgeniy I. Morkovin
Volgograd State Medical University
Email: e.i.morkovin@gmail.com
ORCID iD: 0000-0002-7119-3546
Candidate of Sciences (Medicine), Associate Professor, Head of the Laboratory of Neuropsychopharmacology, Research Center of Innovative Pharma Products
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131Ivan N. Tyurenkov
Volgograd State Medical University
Email: fibfuv@mail.ru
ORCID iD: 0000-0001-7574-3923
Doctor of Sciences (Medicine), Professor, Corresponding Member of the Russian Academy of Sciences, Head of the Laboratory of Pharmacology of Cardiovascular Drugs, Research Center of Innovative Pharma Products, Head of the Department of Pharmacology and Pharmacy, Institute of Continuing Medical and Pharmaceutical Education
Russian Federation, 1, Pavshikh Bortsov Sq., Volgograd, 400131References
- Levine AB, Punihaole D, Levine TB. Characterization of the role of nitric oxide and its clinical applications. Cardiology. 2012;122(1):55–68. doi: 10.1159/000338150.
- Stuart-Smith K. Demystified. Nitric oxide. Mol Pathol. 2002; 55(6):360–6. doi: 10.1136/mp.55.6.360.
- Fukuto JM. A recent history of nitroxyl chemistry, pharmacology and therapeutic potential. Br J Pharmacol. 2019 Jan;176(2):135–46. doi: 10.1111/bph.14384.
- Kourosh-Arami M, Hosseini N, Mohsenzadegan M, Komaki A, Joghataei MT. Neurophysiologic implications of neuronal nitric oxide synthase. Rev Neurosci. 2020 Aug 27;31(6):617–36. doi: 10.1515/revneuro-2019-0111.
- Hardingham N, Dachtler J, Fox K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front Cell Neurosci. 2013 Oct 31;7:190. doi: 10.3389/fncel.2013.00190.
- Dubey H, Gulati K, Ray A. Alzheimer’s Disease: A Contextual Link with Nitric Oxide Synthase. Curr Mol Med. 2020;20(7):505–15. doi: 10.2174/1566524019666191129103117.
- Liskova S. The organ-specific nitric oxide synthase activity in the interaction with sympathetic nerve activity: a hypothesis. Physiol Res. 2021 Apr 30;70(2):169-175. doi: 10.33549/physiolres.934676.
- Fender AC, Dobrev D. Nitric oxide as a fragile switch between cardioprotection and cardiac injury. Int J Cardiol. 2021 Nov 15;343:102–3. doi: 10.1016/j.ijcard.2021.09.001.
- Radziwon-Balicka A, Lesyk G, Back V, Fong T, Loredo-Calderon EL, Dong B, El-Sikhry H, El-Sherbeni AA, El-Kadi A, Ogg S, Siraki A, Seubert JM, Santos-Martinez MJ, Radomski MW, Velazquez-Martinez CA, Winship IR, Jurasz P. Differential eNOS-signalling by platelet subpopulations regulates adhesion and aggregation. Cardiovasc Res. 2017 Dec 1;113(14):1719–31. doi: 10.1093/cvr/cvx179.
- Infante T, Costa D, Napoli C. Novel Insights Regarding Nitric Oxide and Cardiovascular Diseases. Angiology. 2021 May;72(5):411–25. doi: 10.1177/0003319720979243.
- Idrizaj E, Traini C, Vannucchi MG, Baccari MC. Nitric Oxide: From Gastric Motility to Gastric Dysmotility. Int J Mol Sci. 2021 Sep 16;22(18):9990. doi: 10.3390/ijms22189990.
- Hong PP, Zhu XX, Yuan WJ, Niu GJ, Wang JX. Nitric Oxide Synthase Regulates Gut Microbiota Homeostasis by ERK-NF-κB Pathway in Shrimp. Front Immunol. 2021 Dec 3;12:778098. doi: 10.3389/fimmu.2021.778098.
- Sibisi NC, Snyman C, Myburgh KH, Niesler CU. Evaluating the role of nitric oxide in myogenesis in vitro. Biochimie. 2021 Nov 26:S0300-9084(21)00269-8. doi: 10.1016/j.biochi.2021.11.006.
- Balke JE, Zhang L, Percival JM. Neuronal nitric oxide synthase (nNOS) splice variant function: Insights into nitric oxide signaling from skeletal muscle. Nitric Oxide. 2019 Jan 1;82:35–47. doi: 10.1016/j.niox.2018.11.004.
- Baig MS, Zaichick SV, Mao M, de Abreu AL, Bakhshi FR, Hart PC, Saqib U, Deng J, Chatterjee S, Block ML, Vogel SM, Malik AB, Consolaro ME, Christman JW, Minshall RD, Gantner BN, Bonini MG. NOS1-derived nitric oxide promotes NF-κB transcriptional activity through inhibition of suppressor of cytokine signaling-1. J Exp Med. 2015 Sep 21;212(10):1725–38. doi: 10.1084/jem.20140654.
- Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. 2016 Jun 30;2:16045. doi: 10.1038/nrdp.2016.45.
- Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Curr Opin Clin Nutr Metab Care. 2010 Jan;13(1):97–104. doi: 10.1097/MCO.0b013e328332f99d.
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990 Jan;87(2):682–5. doi: 10.1073/pnas.87.2.682.
- Salvemini D, Kim SF, Mollace V. Reciprocal regulation of the nitric oxide and cyclooxygenase pathway in pathophysiology: relevance and clinical implications. Am J Physiol Regul Integr Comp Physiol. 2013 Apr 1;304(7):R473–87. doi: 10.1152/ajpregu.00355.2012.
- Palmieri EM, McGinity C, Wink DA, McVicar DW. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites. 2020 Oct 26;10(11):429. doi: 10.3390/metabo10110429.
- Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012 Apr;33(7):829-37, 837a–837d. doi: 10.1093/eurheartj/ehr304.
- Förstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci U S A. 1991 Mar 1;88(5):1788–92. doi: 10.1073/pnas.88.5.1788.
- Pollock JS, Förstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, Murad F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci USA. 1991 Dec 1;88(23):10480–4. doi: 10.1073/pnas.88.23.10480.
- Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci USA. 1993 Jan 1;90(1):347–51. doi: 10.1073/pnas.90.1.347.
- Sase K, Michel T. Expression of constitutive endothelial nitric oxide synthase in human blood platelets. Life Sci. 1995;57(22):2049–55. doi: 10.1016/0024-3205(95)02191-k.
- Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America. 1994 May;91(10):4214–8. doi: 10.1073/pnas.91.10.4214.
- Marsden PA, Heng HH, Scherer SW, Stewart RJ, Hall AV, Shi XM, Tsui LC, Schappert KT. Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem. 1993 Aug 15;268(23):17478-88.
- Oliveira-Paula GH, Lacchini R, Tanus-Santos JE. Endothelial nitric oxide synthase: From biochemistry and gene structure to clinical implications of NOS3 polymorphisms. Gene. 2016 Jan 10;575(2 Pt 3):584–99. doi: 10.1016/j.gene.2015.09.061.
- Qian J, Fulton D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front Physiol. 2013 Dec 13;4:347. doi: 10.3389/fphys.2013.00347.
- Hall AV, Antoniou H, Wang Y, Cheung AH, Arbus AM, Olson SL, Lu WC, Kau CL, Marsden PA. Structural organization of the human neuronal nitric oxide synthase gene (NOS1). J Biol Chem. 1994 Dec 30;269(52):33082-90.
- Eliasson MJ, Blackshaw S, Schell MJ, Snyder SH. Neuronal nitric oxide synthase alternatively spliced forms: prominent functional localizations in the brain. Proc Natl Acad Sci U S A. 1997 Apr 1;94(7):3396–401. doi: 10.1073/pnas.94.7.3396.
- Sharma NM, Patel KP. Post-translational regulation of neuronal nitric oxide synthase: implications for sympathoexcitatory states. Expert Opin Ther Targets. 2017 Jan;21(1):11–22. doi: 10.1080/14728222.2017.1265505.
- Zhang YH, Jin CZ, Jang JH, Wang Y. Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J Physiol. 2014 Aug 1;592(15):3189–200. doi: 10.1113/jphysiol.2013.270306.
- Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010 Sep 15;23(2):75–93. doi: 10.1016/j.niox.2010.04.007.
- Förstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, Kleinert H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension. 1994 Jun;23(6 Pt 2):1121–31. doi: 10.1161/01.hyp.23.6.1121.
- Tejero J, Shiva S, Gladwin MT. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol Rev. 2019 Jan 1;99(1):311–79. doi: 10.1152/physrev.00036.2017.
- Cinelli MA, Do HT, Miley GP, Silverman RB. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med Res Rev. 2020 Jan;40(1):158–89. doi: 10.1002/med.21599.
- Giulivi C, Poderoso JJ, Boveris A. Production of nitric oxide by mitochondria. J Biol Chem. 1998 May 1;273(18):11038–43. doi: 10.1074/jbc.273.18.11038.
- Tengan CH, Rodrigues GS, Godinho RO. Nitric oxide in skeletal muscle: role on mitochondrial biogenesis and function. Int J Mol Sci. 2012 Dec 14;13(12):17160–84. doi: 10.3390/ijms131217160.
- Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ, Sluse FE, Souza-Pinto NC, Vercesi AE. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal. 2013 Jun 1;18(16):2029–74. doi: 10.1089/ars.2012.4729.
- Tatoyan A, Giulivi C. Purification and characterization of a nitric-oxide synthase from rat liver mitochondria. J Biol Chem. 1998 May 1;273(18):11044–8. doi: 10.1074/jbc.273.18.11044.
- Bates TE, Loesch A, Burnstock G, Clark JB. Immunocytochemical evidence for a mitochondrially located nitric oxide synthase in brain and liver. Biochem Biophys Res Commun. 1995 Aug 24;213(3):896–900. doi: 10.1006/bbrc.1995.2213.
- Gao S, Chen J, Brodsky SV, Huang H, Adler S, Lee JH, Dhadwal N, Cohen-Gould L, Gross SS, Goligorsky MS. Docking of endothelial nitric oxide synthase (eNOS) to the mitochondrial outer membrane: a pentabasic amino acid sequence in the autoinhibitory domain of eNOS targets a proteinase K-cleavable peptide on the cytoplasmic face of mitochondria. J Biol Chem. 2004 Apr 16;279(16):15968–74. doi: 10.1074/jbc.M308504200.
- Elfering SL, Sarkela TM, Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. J Biol Chem. 2002 Oct 11;277(41):38079–86. doi: 10.1074/jbc.M205256200.
- Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci U S A. 2001 Nov 20;98(24):14126–31. doi: 10.1073/pnas.241380298.
- Lacza Z, Snipes JA, Zhang J, Horváth EM, Figueroa JP, Szabó C, Busija DW. Mitochondrial nitric oxide synthase is not eNOS, nNOS or iNOS. Free Radic Biol Med. 2003 Nov 15;35(10):1217–28. doi: 10.1016/s0891-5849(03)00510-0.
- Venkatakrishnan P, Nakayasu ES, Almeida IC, Miller RT. Absence of nitric-oxide synthase in sequentially purified rat liver mitochondria. J Biol Chem. 2009 Jul 24;284(30):19843–55. doi: 10.1074/jbc.M109.003301.
- Parihar MS, Nazarewicz RR, Kincaid E, Bringold U, Ghafourifar P. Association of mitochondrial nitric oxide synthase activity with respiratory chain complex I. Biochem Biophys Res Commun. 2008 Feb 1;366(1):23–8. doi: 10.1016/j.bbrc.2007.11.056.
- Bombicino SS, Iglesias DE, Zaobornyj T, Boveris A, Valdez LB. Mitochondrial nitric oxide production supported by reverse electron transfer. Arch Biochem Biophys. 2016 Oct 1;607:8–19. doi: 10.1016/j.abb.2016.08.010.
- Lacza Z, Pankotai E, Busija DW. Mitochondrial nitric oxide synthase: current concepts and controversies. Front Biosci (Landmark Ed). 2009 Jan 1;14(12):4436–43. doi: 10.2741/3539.
- Giulivi C. Characterization and function of mitochondrial nitric-oxide synthase. Free Radic Biol Med. 2003 Feb 15;34(4):397–408. doi: 10.1016/s0891-5849(02)01298-4.
- Tengan CH, Moraes CT. NO control of mitochondrial function in normal and transformed cells. Biochim Biophys Acta Bioenerg. 2017 Aug;1858(8):573–81. doi: 10.1016/j.bbabio.2017.02.009.
- Shah RC, Sanker S, Wood KC, Durgin BG, Straub AC. Redox regulation of soluble guanylyl cyclase. Nitric Oxide. 2018 Jun 1;76:97–104. doi: 10.1016/j.niox.2018.03.013.
- Sharin VG, Mujoo K, Kots AY, Martin E, Murad F, Sharina IG. Nitric oxide receptor soluble guanylyl cyclase undergoes splicing regulation in differentiating human embryonic cells. Stem Cells Dev. 2011 Jul;20(7):1287–93. doi: 10.1089/scd.2010.0411.
- Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem. 2012;81:533–59. doi: 10.1146/annurev-biochem-050410-100030.
- Montfort WR, Wales JA, Weichsel A. Structure and Activation of Soluble Guanylyl Cyclase, the Nitric Oxide Sensor. Antioxid Redox Signal. 2017 Jan 20;26(3):107–21. doi: 10.1089/ars.2016.6693.
- Förstermann U, Xia N, Li H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ Res. 2017 Feb 17;120(4):713-735. doi: 10.1161/CIRCRESAHA.116.309326.
- Li H, Förstermann U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr Opin Pharmacol. 2013 Apr;13(2):161-7. doi: 10.1016/j.coph.2013.01.006.
- Roe ND, Ren J. Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vascul Pharmacol. 2012 Nov-Dec;57(5-6):168–72. doi: 10.1016/j.vph.2012.02.004.
- Alkaitis MS, Crabtree MJ. Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr Heart Fail Rep. 2012 Sep;9(3):200–10. doi: 10.1007/s11897-012-0097-5.
- Crabtree MJ, Brixey R, Batchelor H, Hale AB, Channon KM. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. J Biol Chem. 2013 Jan 4;288(1):561–9. doi: 10.1074/jbc.M112.415992.
- Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010 Dec 23;468(7327):1115–8. doi: 10.1038/nature09599.
- Chen DD, Chen LY, Xie JB, Shu C, Yang T, Zhou S, Yuan H, Chen AF. Tetrahydrobiopterin regulation of eNOS redox function. Curr Pharm Des. 2014;20(22):3554–62. doi: 10.2174/13816128113196660747.
- Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J Biol Chem. 2009 Oct 9;284(41):28128–36. doi: 10.1074/jbc.M109.041483.
- Kietadisorn R, Kietselaer BL, Schmidt HH, Moens AL. Role of tetrahydrobiopterin (BH4) in hyperhomocysteinemia-induced endothelial dysfuction: new indication for this orphan-drug? Am J Physiol Endocrinol Metab. 2011 Jun;300(6):E1176; author reply E1177-8. doi: 10.1152/ajpendo.00084.2011.
- Dikalova A, Aschner JL, Kaplowitz MR, Summar M, Fike CD. Tetrahydrobiopterin oral therapy recouples eNOS and ameliorates chronic hypoxia-induced pulmonary hypertension in newborn pigs. Am J Physiol Lung Cell Mol Physiol. 2016 Oct 1;311(4):L743–L753. doi: 10.1152/ajplung.00238.2016.
- Suckling CJ, Gibson CL, Huggan JK, Morthala RR, Clarke B, Kununthur S, Wadsworth RM, Daff S, Papale D. 6-Acetyl-7,7-dimethyl-5,6,7,8-tetrahydropterin is an activator of nitric oxide synthases. Bioorg Med Chem Lett. 2008 Mar 1;18(5):1563–6. doi: 10.1016/j.bmcl.2008.01.079.
- An H, Wei R, Ke J, Yang J, Liu Y, Wang X, Wang G, Hong T. Metformin attenuates fluctuating glucose-induced endothelial dysfunction through enhancing GTPCH1-mediated eNOS recoupling and inhibiting NADPH oxidase. J Diabetes Complications. 2016 Aug;30(6):1017–24. doi: 10.1016/j.jdiacomp.2016.04.018.
- Caldwell RB, Toque HA, Narayanan SP, Caldwell RW. Arginase: an old enzyme with new tricks. Trends Pharmacol Sci. 2015 Jun;36(6):395–405. doi: 10.1016/j.tips.2015.03.006.
- Caldwell RW, Rodriguez PC, Toque HA, Narayanan SP, Caldwell RB. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol Rev. 2018 Apr 1;98(2):641–65. doi: 10.1152/physrev.00037.2016.
- Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol (1985). 2009 Oct;107(4):1249–57. doi: 10.1152/japplphysiol.91393.2008.
- El-Bassossy HM, El-Fawal R, Fahmy A, Watson ML. Arginase inhibition alleviates hypertension in the metabolic syndrome. Br J Pharmacol. 2013 Jun;169(3):693–703. doi: 10.1111/bph.12144.
- Polis B, Srikanth KD, Gurevich V, Gil-Henn H, Samson AO. L-Norvaline, a new therapeutic agent against Alzheimer’s disease. Neural Regen Res. 2019 Sep;14(9):1562–72. doi: 10.4103/1673-5374.255980.
- Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005 Dec 9;97(12):1232–5. doi: 10.1161/01.RES.0000196564.18314.23.
- Rohilla A, Rohilla S, Kumar A, Khan MU, Deep A. Pleiotropic effects of statins: A boulevard to cardioprotection. Arab J Chem. 2016; 9. – P. S21–S27. doi: 10.1016/j.arabjc.2011.06.025.
- Gorabi AM, Kiaie N, Hajighasemi S, Banach M, Penson PE, Jamialahmadi T, Sahebkar A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J Clin Med. 2019 Nov 22;8(12):2051. doi: 10.3390/jcm8122051.
- Margaritis M, Channon KM, Antoniades C. Statins as regulators of redox state in the vascular endothelium: beyond lipid lowering. Antioxid Redox Signal. 2014 Mar 10;20(8):1198–215. doi: 10.1089/ars.2013.5430.
- Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, Van-Assche T, Cunnington C, Tousoulis D, Pillai R, Ratnatunga C, Stefanadis C, Channon KM. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J. 2009 May;30(9):1142–50. doi: 10.1093/eurheartj/ehp061.
- Serban C, Sahebkar A, Ursoniu S, Mikhailidis DP, Rizzo M, Lip GY, Kees Hovingh G, Kastelein JJ, Kalinowski L, Rysz J, Banach MA. A systematic review and meta-analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Sci Rep. 2015; 5(1): 9902. doi: 10.1038/srep09902.
- Xia N, Förstermann U, Li H. Resveratrol and endothelial nitric oxide. Molecules. 2014 Oct 9;19(10):16102–21. doi: 10.3390/molecules191016102.
- Suschek C, Kolb H, Kolb-Bachofen V. Dobesilate enhances endothelial nitric oxide synthase-activity in macro- and microvascular endothelial cells. Br J Pharmacol. 1997 Dec;122(7):1502–8. doi: 10.1038/sj.bjp.0701512.
- Carrizzo A, Lenzi P, Procaccini C, Damato A, Biagioni F, Ambrosio M, Amodio G, Remondelli P, Del Giudice C, Izzo R, Malovini A, Formisano L, Gigantino V, Madonna M, Puca AA, Trimarco B, Matarese G, Fornai F, Vecchione C. Pentraxin 3 Induces Vascular Endothelial Dysfunction Through a P-selectin/Matrix Metalloproteinase-1 Pathway. Circulation. 2015 Apr 28;131(17):1495–505. doi: 10.1161/CIRCULATIONAHA.114.014822.
- Leung WK, Gao L, Siu PM, Lai CW. Diabetic nephropathy and endothelial dysfunction: Current and future therapies, and emerging of vascular imaging for preclinical renal-kinetic study. Life Sci. 2016 Dec 1;166:121–30. doi: 10.1016/j.lfs.2016.10.015.
- Zhou Y, Qi C, Li S, Shao X, Ni Z. Investigation of the Mechanism Underlying Calcium Dobesilate-Mediated Improvement of Endothelial Dysfunction and Inflammation Caused by High Glucose. Mediators Inflamm. 2019; 2019: 9893682. doi: 10.1155/2019/9893682.
- Sison K, Eremina V, Baelde H, Min W, Hirashima M, Fantus IG, Quaggin SE. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010 Oct;21(10):1691–701. doi: 10.1681/ASN.2010030295.
- Baelde HJ, Eikmans M, Lappin DW, Doran PP, Hohenadel D, Brinkkoetter PT, van der Woude FJ, Waldherr R, Rabelink TJ, de Heer E, Bruijn JA. Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int. 2007 Apr;71(7):637–45. doi: 10.1038/sj.ki.5002101.
- Haller H, Ji L, Stahl K, Bertram A, Menne J. Molecular Mechanisms and Treatment Strategies in Diabetic Nephropathy: New Avenues for Calcium Dobesilate-Free Radical Scavenger and Growth Factor Inhibition. Biomed Res Int. 2017;2017:1909258. doi: 10.1155/2017/1909258.
- Kraehling JR, Sessa WC. Contemporary Approaches to Modulating the Nitric Oxide-cGMP Pathway in Cardiovascular Disease. Circ Res. 2017 Mar 31;120(7):1174–82. doi: 10.1161/CIRCRESAHA.117.303776.
- Hou HT, Wang J, Zhang X, Wang ZQ, Chen TN, Zhang JL, Yang Q, He GW. Endothelial nitric oxide synthase enhancer AVE3085 reverses endothelial dysfunction induced by homocysteine in human internal mammary arteries. Nitric Oxide. 2018; 81:21–7. doi: 10.1016/j.niox.2018.10.001.
- White AR, Ryoo S, Bugaj L, Attarzadeh DO, Thiyagarajan S, Chen K, Attwater S, Abbot B, Li D, Champion HC, Shoukas AA, Nyhan D, Hare JM, Berkowitz DE, Tuday EC. Early changes in vasoreactivity after simulated microgravity are due to an upregulation of the endothelium-dependent nitric oxide/cGMP pathway. Eur J Appl Physiol. 2010 Sep;110(2):395–404. doi: 10.1007/s00421-010-1514-7.
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001 Aug 1;357(Pt 3):593–615. doi: 10.1042/0264-6021:3570593.
- Schulman IH, Hare JM. Regulation of cardiovascular cellular processes by S-nitrosylation. Biochim Biophys Acta. 2012 Jun;1820(6):752–62. doi: 10.1016/j.bbagen.2011.04.002.
- Poulos TL, Li H. Nitric oxide synthase and structure-based inhibitor design. Nitric Oxide. 2017 Feb 28;63:68–77. doi: 10.1016/j.niox.2016.11.004.
- Wong VW, Lerner E. Nitric oxide inhibition strategies. Future Sci OA. 2015; 1(1): FSO35. doi: 10.4155/fso.15.35.
- Víteček J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. 2012;2012:318087. doi: 10.1155/2012/318087.
- Park J, Pramanick S, Park D, Yeo J, Lee J, Lee H, Kim WJ. Therapeutic-Gas-Responsive Hydrogel. Adv Mater. 2017 Nov;29(44). doi: 10.1002/adma.201702859.
- Korhonen R, Lahti A, Hämäläinen M, Kankaanranta H, Moilanen E. Dexamethasone inhibits inducible nitric-oxide synthase expression and nitric oxide production by destabilizing mRNA in lipopolysaccharide-treated macrophages. Mol Pharmacol. 2002 Sep;62(3):698–704. doi: 10.1124/mol.62.3.698.
- Yeo J, Lee YM, Lee J, Park D, Kim K, Kim J, Park J, Kim WJ. Nitric Oxide-Scavenging Nanogel for Treating Rheumatoid Arthritis. Nano Lett. 2019 Oct 9;19(10):6716–24. doi: 10.1021/acs.nanolett.9b00496.
- Putzke J, Seidel B, Huang PL, Wolf G. Differential expression of alternatively spliced isoforms of neuronal nitric oxide synthase (nNOS) and N-methyl-D-aspartate receptors (NMDAR) in knockout mice deficient in nNOS alpha (nNOS alpha(Delta/Delta) mice). Brain Res Mol Brain Res. 2000 Dec 28;85(1-2):13–23. doi: 10.1016/s0169-328x(00)00220-5.
- Frandsen U, Lopez-Figueroa M, Hellsten Y. Localization of nitric oxide synthase in human skeletal muscle. Biochem Biophys Res Commun. 1996 Oct 3;227(1):88–93. doi: 10.1006/bbrc.1996.1472.
- El-Yazbi AF, Cho WJ, Cena J, Schulz R, Daniel EE. Smooth muscle NOS, colocalized with caveolin-1, modulates contraction in mouse small intestine. J Cell Mol Med. 2008 Aug;12(4):1404–15. doi: 10.1111/j.1582-4934.2008.00335.x.
- Saluja R, Saini R, Mitra K, Bajpai VK, Dikshit M. Ultrastructural immunogold localization of nitric oxide synthase isoforms in rat and human eosinophils. Cell Tissue Res. 2010 May;340(2):381–8. doi: 10.1007/s00441-010-0947-y.
- Saini R, Singh S. Inducible nitric oxide synthase: An asset to neutrophils. J Leukoc Biol. 2019 Jan;105(1):49–61. doi: 10.1002/JLB.4RU0418-161R.
- Feron O, Dessy C, Moniotte S, Desager JP, Balligand JL. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J Clin Invest. 1999 Mar;103(6):897–905. doi: 10.1172/JCI4829.
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008 Jun;27(11):2783–802. doi: 10.1111/j.1460-9568.2008.06285.x.
- Lajoix AD, Reggio H, Chardès T, Péraldi-Roux S, Tribillac F, Roye M, Dietz S, Broca C, Manteghetti M, Ribes G, Wollheim CB, Gross R. A neuronal isoform of nitric oxide synthase expressed in pancreatic beta-cells controls insulin secretion. Diabetes. 2001 Jun;50(6):1311–23. doi: 10.2337/diabetes.50.6.1311.
- Schwarz PM, Kleinert H, Förstermann U. Potential functional significance of brain-type and muscle-type nitric oxide synthase I expressed in adventitia and media of rat aorta. Arterioscler Thromb Vasc Biol. 1999 Nov;19(11):2584–90. doi: 10.1161/01.atv.19.11.2584.
- Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004 Oct 1;500(1-3):255–66. doi: 10.1016/j.ejphar.2004.07.030.
- Förstermann U, Schmidt HH, Pollock JS, Sheng H, Mitchell JA, Warner TD, Nakane M, Murad F. Isoforms of nitric oxide synthase. Characterization and purification from different cell types. Biochem Pharmacol. 1991 Oct 24;42(10):1849–57. doi: 10.1016/0006-2952(91)90581-o.
- Dombernowsky NW, Ölmestig JNE, Witting N, Kruuse C. Role of neuronal nitric oxide synthase (nNOS) in Duchenne and Becker muscular dystrophies – Still a possible treatment modality? Neuromuscul Disord. 2018 Nov;28(11):914–26. doi: 10.1016/j.nmd.2018.09.001.
- Mörlin B, Andersson E, Byström B, Hammarström M. Nitric oxide induces endometrial secretion at implantation time. Acta Obstet Gynecol Scand. 2005 Nov;84(11):1029–34. doi: 10.1111/j.0001-6349.2005.00804.x.
- Lu D, Fu Y, Lopez-Ruiz A, Zhang R, Juncos R, Liu H, Manning RD Jr, Juncos LA, Liu R. Salt-sensitive splice variant of nNOS expressed in the macula densa cells. Am J Physiol Renal Physiol. 2010 Jun;298(6):F1465–71. doi: 10.1152/ajprenal.00650.2009.
- Förstermann U, Li H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br J Pharmacol. 2011 Sep;164(2):213–23. doi: 10.1111/j.1476-5381.2010.01196.x.
- Moncada S. Nitric oxide: discovery and impact on clinical medicine. J R Soc Med. 1999 Apr;92(4):164–9. doi: 10.1177/014107689909200402.
- Tojo A, Onozato ML, Fujita T. Role of macula densa neuronal nitric oxide synthase in renal diseases. Med Mol Morphol. 2006 Mar;39(1):2–7. doi: 10.1007/s00795-006-0310-2.
- Takahashi T. Pathophysiological significance of neuronal nitric oxide synthase in the gastrointestinal tract. J Gastroenterol. 2003;38(5):421–30. doi: 10.1007/s00535-003-1094-y.
- Xu L, Xie K, Fidler IJ. Therapy of human ovarian cancer by transfection with the murine interferon beta gene: role of macrophage-inducible nitric oxide synthase. Hum Gene Ther. 1998 Dec 10;9(18):2699–708. doi: 10.1089/hum.1998.9.18-2699.
- Kröncke KD, Fehsel K, Kolb-Bachofen V. Inducible nitric oxide synthase and its product nitric oxide, a small molecule with complex biological activities. Biol Chem Hoppe Seyler. 1995 Jun;376(6):327–43. doi: 10.1515/bchm3.1995.376.6.327.
- Moncada S, Higgs EA. Endogenous nitric oxide: physiology, pathology and clinical relevance. Eur J Clin Invest. 1991 Aug;21(4):361–74. doi: 10.1111/j.1365-2362.1991.tb01383.x.
- Langrehr JM, Hoffman RA, Lancaster JR Jr, Simmons RL. Nitric oxide--a new endogenous immunomodulator. Transplantation. 1993 Jun;55(6):1205–12. doi: 10.1097/00007890-199306000-00001.
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003 Nov;9(11):1370–6. doi: 10.1038/nm948.
- Zhou QG, Zhu XH, Nemes AD, Zhu DY. Neuronal nitric oxide synthase and affective disorders. IBRO Rep. 2018 Nov 17;5:116–32. doi: 10.1016/j.ibror.2018.11.004.
Supplementary files

